Introduction
Acute flaccid paralysis (AFP) is a serious medical emergency characterized by the rapid onset of weakness and loss of muscle tone. Primarily affecting children, but also observed in adults, AFP presents a significant diagnostic challenge due to its diverse etiologies, ranging from infectious agents to immune-mediated and structural disorders. Since 2012, the global landscape of AFP has been further complicated by outbreaks of acute flaccid myelitis (AFM), a polio-like illness often linked to non-polio enteroviruses. The clinical urgency of AFP stems from its potential to cause severe disability, respiratory failure, and even death. Distinguishing between the various causes of AFP is paramount for timely and appropriate management, influencing both acute interventions and long-term rehabilitation strategies. This review aims to provide a comprehensive guide to the differential diagnosis of acute flaccid paralysis, drawing upon the clinical features, epidemiological context, and diagnostic modalities necessary for accurate differentiation. Understanding the nuances of AFP and its mimics is crucial for healthcare professionals to navigate this complex clinical scenario effectively, ensuring optimal patient outcomes.
Understanding Acute Flaccid Paralysis (AFP)
Acute flaccid paralysis is not a disease in itself but rather a clinical syndrome, defined by the sudden or rapid onset of weakness (paresis) or complete paralysis (plegia) accompanied by decreased muscle tone (flaccidity) and reduced or absent reflexes (hypo- or areflexia). The acute nature of onset, typically developing over hours to a few days, necessitates prompt medical evaluation to identify the underlying cause and initiate appropriate treatment. AFP is a critical clinical presentation due to its potential to indicate severe neurological compromise, often involving the anterior horn cells of the spinal cord or peripheral nerves, which are essential for motor function.
The importance of differential diagnosis in AFP cannot be overstated. The syndrome can be triggered by a wide spectrum of conditions, each requiring distinct management strategies. For instance, while supportive care is central to managing AFM, conditions like Guillain-Barré syndrome (GBS) may benefit from immunomodulatory therapies such as intravenous immunoglobulin (IVIG) or plasma exchange. Similarly, identifying and treating infectious causes like poliomyelitis or West Nile virus is fundamentally different from managing non-infectious etiologies such as spinal cord infarction or periodic paralysis. Misdiagnosis or delayed diagnosis can lead to suboptimal treatment, potentially exacerbating neurological damage and long-term disability. Therefore, a systematic approach to differential diagnosis is essential for clinicians encountering patients with AFP. This approach must integrate clinical assessment, epidemiological considerations, neuroimaging, electrophysiological studies, and laboratory investigations to narrow down the diagnostic possibilities and guide targeted interventions.
Epidemiology and Etiology of AFP
The epidemiology of acute flaccid paralysis is intricately linked to its diverse range of causes. Globally, the most historically significant cause of AFP is poliomyelitis, caused by poliovirus. However, due to the success of global polio eradication efforts, wild-type poliovirus is now rare, although vaccine-derived poliovirus remains a concern in some regions with low vaccination coverage. The focus has shifted towards non-polio enteroviruses, particularly enterovirus D68 (EV-D68) and enterovirus A71 (EV-A71), as significant contributors to AFP, especially in the context of AFM outbreaks observed since 2012.
AFM outbreaks have demonstrated a seasonal, often biennial pattern, particularly in temperate regions, typically peaking in the late summer and fall. This seasonality, coupled with prodromal respiratory illnesses reported in many AFM cases, strongly suggests a viral etiology. Enteroviruses, known for their neurotropic potential, have been implicated as the primary culprits. EV-D68 has been most prominently associated with AFM outbreaks in North America and Europe, while EV-A71 is more frequently linked to AFP cases in the Asia-Pacific region. However, it’s crucial to note that not all cases of AFP are attributable to enteroviruses.
Beyond enteroviruses, other infectious agents can cause AFP, including West Nile virus, Japanese encephalitis virus, and tick-borne encephalitis virus, particularly in endemic areas. These arboviral infections often present with AFP alongside systemic symptoms and meningoencephalitis. Bacterial infections, such as certain strains of Campylobacter jejuni, are well-established triggers for Guillain-Barré syndrome, a major differential diagnosis of AFP.
Non-infectious causes of AFP are equally important to consider. Guillain-Barré syndrome, an autoimmune polyneuropathy, is a leading cause of AFP worldwide. Transverse myelitis, encompassing demyelinating conditions like neuromyelitis optica spectrum disorder (NMOSD) and myelin oligodendrocyte glycoprotein antibody-associated disease (MOGAD), as well as idiopathic transverse myelitis, can manifest with AFP. Spinal cord infarction, although less common in children, represents a vascular etiology of AFP. Other rarer causes include periodic paralysis, botulism, toxic neuropathies, and certain metabolic disorders.
Understanding the epidemiological context is vital for differential diagnosis. Factors such as geographic location, season, age of the patient, vaccination history, and recent travel can provide crucial clues towards the likely etiology of AFP. For instance, in regions where wild poliovirus persists, AFP cases warrant immediate investigation for poliomyelitis. During enterovirus outbreak seasons, AFM becomes a higher diagnostic consideration, especially in children presenting with prodromal respiratory illness and asymmetric limb weakness. In endemic areas, arboviral infections should be considered in patients with AFP and accompanying systemic or encephalitic features. Therefore, a thorough epidemiological assessment is an indispensable first step in approaching AFP, guiding subsequent diagnostic investigations and narrowing the differential diagnosis.
Clinical Presentation: Key Features and Red Flags for Differential Diagnosis
The clinical presentation of acute flaccid paralysis is characterized by the rapid onset of muscle weakness, but the specific features can vary significantly depending on the underlying etiology. Recognizing subtle differences in clinical presentation is crucial for navigating the differential diagnosis.
Weakness Pattern: The distribution and pattern of weakness are highly informative. AFM typically presents with asymmetric weakness, often affecting one or more limbs, with a predilection for the upper limbs and proximal muscle groups. In contrast, Guillain-Barré syndrome often manifests with symmetric, ascending weakness, starting distally in the legs and progressing upwards. Spinal cord infarction tends to cause symmetric weakness below the level of the lesion. Focal weakness limited to cranial nerves without limb involvement is less typical for AFM but can occur, expanding the differential to include brainstem lesions of various etiologies.
Sensory Involvement: Sensory symptoms play a critical role in differentiating AFP causes. Guillain-Barré syndrome is often associated with prominent sensory symptoms such as paresthesia, numbness, and pain, although the acute motor axonal neuropathy (AMAN) variant may have minimal sensory findings. In AFM, sensory symptoms are generally mild and non-specific, such as transient paresthesia or neuropathic pain. Transverse myelitis frequently presents with a distinct sensory level, indicating the level of spinal cord involvement, along with bowel and bladder dysfunction. Spinal cord infarction can also present with a sensory level and severe pain at onset.
Reflexes and Muscle Tone: Flaccidity and areflexia or hyporeflexia are hallmark features of AFP, reflecting lower motor neuron involvement. However, the degree and distribution can offer diagnostic hints. In AFM, affected limbs are typically flaccid and areflexic. Guillain-Barré syndrome also presents with areflexia, which is often generalized. In upper motor neuron lesions, such as in early spinal shock from transverse myelitis or spinal cord infarction, flaccidity and areflexia may initially be present, but can evolve into spasticity and hyperreflexia over time.
Associated Symptoms: Prodromal symptoms are common in infectious etiologies of AFP. AFM is frequently preceded by a febrile illness with respiratory symptoms. Guillain-Barré syndrome often follows a respiratory or gastrointestinal infection. Arboviral infections may present with fever, rash, headache, and altered mental status. The presence or absence of fever, rash, meningeal signs, or encephalopathy can help differentiate between infectious and non-infectious causes.
Pain: Pain is a variable feature in AFP. Limb or back pain can precede weakness in AFM. Neuropathic pain is a prominent feature of Guillain-Barré syndrome. Severe, sudden onset back or limb pain is characteristic of spinal cord infarction. The nature, onset, and location of pain can contribute to differential diagnosis.
Bulbar and Cranial Nerve Involvement: Bulbar weakness (affecting swallowing, speech, and airway protection) and facial weakness are common in both AFM and Guillain-Barré syndrome. However, the specific cranial nerve involvement can differ. AFM may involve a range of cranial nerves, including facial, bulbar, and extraocular muscles. Guillain-Barré syndrome often presents with bilateral facial weakness and bulbar dysfunction. Botulism, another differential diagnosis, characteristically presents with descending paralysis, often starting with cranial nerve palsies, including ptosis, diplopia, and bulbar weakness.
Rate of Progression and Nadir: The temporal profile of weakness progression is diagnostically relevant. AFM typically reaches its nadir of weakness within hours to days. Guillain-Barré syndrome progresses over days to weeks. Spinal cord infarction reaches maximal deficit within minutes to hours. Transverse myelitis evolves over days to weeks. The time course of weakness development helps narrow the diagnostic possibilities.
Red Flags for Alternative Diagnoses: Certain clinical features should raise suspicion for diagnoses other than AFM and prompt consideration of alternative etiologies. Encephalopathy, significant sensory deficits, bowel and bladder dysfunction, and upper motor neuron signs (spasticity, hyperreflexia) are less typical for AFM and more suggestive of conditions like transverse myelitis, spinal cord infarction, or encephalomyelitis. A history of previous neurological events, optic neuritis, or clinical evolution extending beyond 10 days also points away from AFM towards demyelinating conditions.
By carefully evaluating the pattern of weakness, sensory findings, reflexes, associated symptoms, pain characteristics, cranial nerve involvement, and the temporal profile of illness, clinicians can begin to differentiate between the various causes of AFP based on clinical presentation alone. This clinical assessment forms the foundation for subsequent targeted investigations to confirm the diagnosis.
Differential Diagnosis of Acute Flaccid Paralysis
The differential diagnosis of acute flaccid paralysis is broad and requires a systematic approach to narrow down possibilities based on clinical, epidemiological, and investigative findings. Key conditions to consider include:
1. Acute Flaccid Myelitis (AFM):
- Etiology: Primarily associated with non-polio enteroviruses, especially EV-D68.
- Clinical Features: Predominantly in children, prodromal febrile illness with respiratory symptoms, acute onset asymmetric limb weakness (arms > legs), flaccidity, hyporeflexia, cranial nerve involvement (bulbar, facial), minimal sensory findings.
- Diagnostic Clues: Seasonal occurrence, MRI showing spinal cord gray matter lesions (T2 hyperintensity, anterior horn predominance), CSF pleocytosis (mild to moderate).
- Differentiating Features: Asymmetric weakness, gray matter spinal cord lesions, prodromal respiratory illness, less prominent sensory findings compared to mimics.
2. Guillain-Barré Syndrome (GBS):
- Etiology: Autoimmune polyneuropathy, often post-infectious (e.g., Campylobacter jejuni, viral infections).
- Clinical Features: Symmetric, ascending weakness (legs > arms), areflexia, prominent sensory symptoms (paresthesia, pain), bulbar and facial weakness, autonomic dysfunction.
- Diagnostic Clues: Albuminocytologic dissociation in CSF (elevated protein, normal cell count), nerve conduction studies showing demyelination or axonal neuropathy.
- Differentiating Features: Symmetric weakness, ascending pattern, prominent sensory involvement, albuminocytologic dissociation, nerve conduction study findings.
3. Transverse Myelitis (TM):
- Etiology: Inflammation of the spinal cord, can be idiopathic, demyelinating (NMOSD, MOGAD, multiple sclerosis), or post-infectious.
- Clinical Features: Rapid onset weakness, sensory level, bowel and bladder dysfunction, back pain, can be symmetric or asymmetric, may have upper motor neuron signs (spasticity, hyperreflexia) depending on stage.
- Diagnostic Clues: MRI showing spinal cord lesions (often longitudinally extensive, can involve white and gray matter), CSF pleocytosis (variable), serum antibodies (AQP4-IgG, MOG-IgG) in demyelinating subtypes.
- Differentiating Features: Sensory level, bowel/bladder dysfunction, potential for upper motor neuron signs, longitudinally extensive spinal cord lesions, antibody testing for demyelinating subtypes.
4. Spinal Cord Infarction:
- Etiology: Vascular occlusion of spinal arteries, often anterior spinal artery.
- Clinical Features: Sudden onset, severe back or limb pain at onset (“knife-like”), symmetric weakness below the level of infarction, sensory level, bowel and bladder dysfunction, rapid progression to nadir (minutes to hours).
- Diagnostic Clues: MRI showing spinal cord infarction pattern (often “owl-eyes” appearance on axial T2), lack of gadolinium enhancement, angiography may be considered.
- Differentiating Features: Abrupt onset with severe pain, rapid nadir, symmetric weakness, characteristic MRI findings of infarction.
5. Poliomyelitis:
- Etiology: Poliovirus infection (wild-type or vaccine-derived).
- Clinical Features: Asymmetric flaccid paralysis, fever, muscle pain, bulbar weakness, historically more common in unvaccinated populations.
- Diagnostic Clues: Stool viral culture and PCR for poliovirus, epidemiological context (travel to endemic areas, vaccination status).
- Differentiating Features: Asymmetric paralysis, stool poliovirus detection, epidemiological context, now rare due to eradication efforts.
6. West Nile Virus Myelitis:
- Etiology: West Nile virus infection, transmitted by mosquitoes.
- Clinical Features: Fever, rash, headache, myalgia, encephalitis, asymmetric flaccid paralysis.
- Diagnostic Clues: Serology for West Nile virus, CSF analysis, MRI may show spinal cord and brain involvement, endemic area exposure.
- Differentiating Features: Seasonal occurrence in endemic areas, rash, encephalitis features, West Nile virus serology.
7. Japanese Encephalitis:
- Etiology: Japanese encephalitis virus, transmitted by mosquitoes.
- Clinical Features: Fever, headache, altered mental status, seizures, tremors, movement disorders, flaccid paralysis (less common than encephalitic features).
- Diagnostic Clues: Travel to endemic regions in Asia, serology for Japanese encephalitis virus, MRI showing basal ganglia and thalamic involvement, CSF analysis.
- Differentiating Features: Travel history, encephalitic presentation, basal ganglia involvement on MRI, Japanese encephalitis serology.
8. Tick-borne Encephalitis:
- Etiology: Tick-borne encephalitis virus, transmitted by ticks.
- Clinical Features: Fever, headache, meningeal signs, encephalitis, flaccid paralysis (less common).
- Diagnostic Clues: Tick exposure history, travel to endemic regions in Europe and Asia, serology for tick-borne encephalitis virus, CSF analysis.
- Differentiating Features: Tick exposure, geographical context, encephalitic presentation, tick-borne encephalitis serology.
9. Botulism:
- Etiology: Clostridium botulinum toxin, foodborne, wound, or infant botulism.
- Clinical Features: Descending paralysis (cranial nerves first: ptosis, diplopia, bulbar weakness, then limb weakness), symmetric weakness, dilated pupils, dry mouth, constipation, preserved sensation.
- Diagnostic Clues: Electrophysiological studies (incremental response on repetitive nerve stimulation), toxin assay in serum or stool, history of canned food ingestion or wound infection.
- Differentiating Features: Descending paralysis, cranial nerve involvement, autonomic findings, electrophysiological features.
10. Periodic Paralysis:
- Etiology: Genetic mutations in ion channel genes, hypokalemic, hyperkalemic, or normokalemic forms.
- Clinical Features: Episodic weakness, often triggered by rest after exercise, carbohydrate-rich meals, or potassium fluctuations, symmetric weakness, no sensory findings, normal reflexes between attacks.
- Diagnostic Clues: Serum potassium levels during attacks, family history, genetic testing.
- Differentiating Features: Episodic nature of weakness, potassium levels during attacks, family history.
11. Toxic Neuropathies:
- Etiology: Exposure to toxins (e.g., organophosphates, heavy metals).
- Clinical Features: Variable presentation depending on toxin, can include flaccid paralysis, sensory and autonomic symptoms, history of exposure.
- Diagnostic Clues: Detailed history of exposure, specific toxin testing (e.g., cholinesterase levels in organophosphate poisoning).
- Differentiating Features: Exposure history, specific toxin-related symptoms, relevant laboratory tests.
This comprehensive list highlights the breadth of differential diagnoses for AFP. A structured diagnostic approach is essential to systematically evaluate each possibility and arrive at an accurate diagnosis.
Diagnostic Approach to AFP: A Step-by-Step Guide for Clinicians
The diagnostic approach to acute flaccid paralysis requires a stepwise strategy, integrating clinical assessment with targeted investigations to differentiate between the various etiologies. A practical algorithm involves the following steps:
1. Initial Assessment and History:
- Detailed History: Obtain a thorough history, including the onset, progression, and pattern of weakness, associated symptoms (fever, rash, sensory changes, pain, bowel/bladder dysfunction), prodromal illness, vaccination history (especially polio), travel history, exposure risks (tick bites, mosquito exposure, food history), family history of neurological disorders, and medications.
- Neurological Examination: Perform a comprehensive neurological exam, focusing on muscle strength (using MRC scale), tone, reflexes, sensory function, cranial nerves, and meningeal signs. Document the distribution and severity of weakness, presence of asymmetry, sensory level, and any upper motor neuron signs.
2. Neuroimaging: MRI of the Spinal Cord:
- Spinal MRI: MRI of the entire spinal cord is the most crucial neuroimaging study in AFP. It helps to identify spinal cord lesions, their location (gray vs. white matter), extent (longitudinal), and characteristics (edema, enhancement).
- AFM: Characteristic gray matter T2 hyperintensity, often anterior horns, longitudinally extensive, minimal or no enhancement.
- Transverse Myelitis: Spinal cord lesion, can involve both gray and white matter, often longitudinally extensive, may show enhancement.
- Spinal Cord Infarction: “Owl-eyes” appearance on axial T2, anterior cord involvement, lack of enhancement.
- Guillain-Barré Syndrome: Spinal cord MRI is typically normal or may show nerve root enhancement, but the cord parenchyma is usually normal.
3. Cerebrospinal Fluid (CSF) Analysis:
- Lumbar Puncture: CSF analysis is essential to evaluate for inflammation, infection, and other abnormalities.
- AFM: Mild to moderate pleocytosis (lymphocytic predominance), normal to mildly elevated protein.
- Guillain-Barré Syndrome: Albuminocytologic dissociation (elevated protein, normal or mildly elevated cell count).
- Transverse Myelitis: Pleocytosis (variable), elevated protein, oligoclonal bands may be present in demyelinating TM.
- Spinal Cord Infarction: CSF may be normal or show mild pleocytosis and elevated protein.
- Infectious Myelitis (Viral, Bacterial, Fungal): Pleocytosis, elevated protein, glucose may be low in bacterial/fungal infections, pathogen-specific tests (viral PCR, bacterial culture, fungal stains).
4. Electrophysiological Studies: EMG/NCS:
- Electromyography (EMG) and Nerve Conduction Studies (NCS): These studies help differentiate between neuropathic, myopathic, and neuromuscular junction disorders.
- AFM: Motor neuronopathy pattern, reduced CMAPs, denervation potentials (fibrillations, positive sharp waves).
- Guillain-Barré Syndrome: Demyelinating or axonal neuropathy patterns (slowed conduction velocities, conduction block, reduced CMAPs).
- Botulism: Incremental response on repetitive nerve stimulation.
- Periodic Paralysis: Normal EMG/NCS between attacks, abnormal during attacks (reduced CMAPs).
5. Targeted Laboratory Investigations:
- Infectious Etiologies:
- Viral PCR: Respiratory and stool samples for enteroviruses (EV-D68, EV-A71) in suspected AFM, CSF for enteroviruses (low yield), West Nile virus, Japanese encephalitis virus, tick-borne encephalitis virus.
- Serology: West Nile virus, Japanese encephalitis virus, tick-borne encephalitis virus, poliovirus (if indicated).
- Bacterial Culture and Serology: Campylobacter jejuni serology in suspected GBS.
- Botulism Toxin Assay: Serum and stool for botulinum toxin in suspected botulism.
- Immune-Mediated and Demyelinating Conditions:
- Serum Antibodies: AQP4-IgG and MOG-IgG for NMOSD and MOGAD in suspected transverse myelitis, anti-ganglioside antibodies (GM1, GD1a, etc.) in suspected GBS (variable sensitivity and specificity).
- Metabolic and Genetic Disorders:
- Serum Electrolytes: Potassium levels during attacks of suspected periodic paralysis.
- Genetic Testing: For periodic paralysis if suspected.
- Toxic Exposures:
- Toxicology Screening: If toxic neuropathy is suspected, based on history.
6. Epidemiological and Contextual Assessment:
- Epidemiological Data: Consider local epidemiology of AFP causes, seasonal patterns, outbreaks, and regional prevalence of specific pathogens (e.g., West Nile virus in North America, Japanese encephalitis in Asia).
- Risk Factors: Evaluate risk factors for specific conditions, such as vaccination status for polio, travel to endemic areas for arboviruses, dietary history for botulism, and family history for periodic paralysis.
7. Clinical Follow-up and Re-evaluation:
- Serial Examinations: Monitor the clinical course, progression or improvement of weakness, and evolution of neurological signs.
- Repeat Investigations: Repeat MRI or CSF analysis if initial studies are inconclusive or if the clinical picture evolves.
- Response to Treatment: Assess response to empiric or targeted therapies, which can further refine the diagnosis.
By systematically following these steps, clinicians can effectively narrow the differential diagnosis of AFP, guide appropriate investigations, and initiate timely and targeted management strategies. Accurate and rapid diagnosis is crucial to improve outcomes and minimize long-term disability in patients with acute flaccid paralysis.
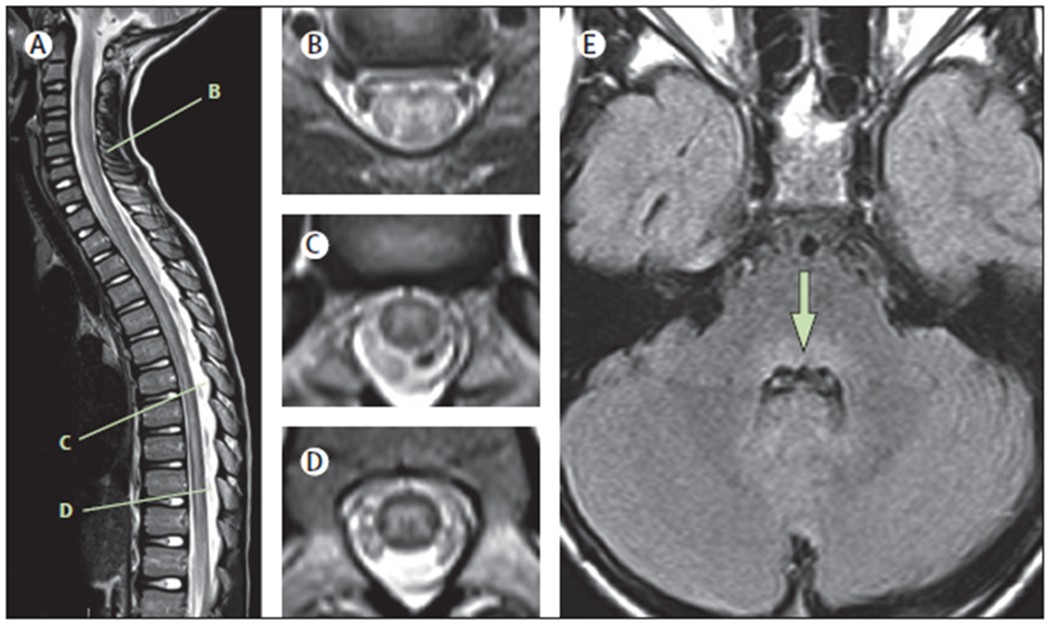
Figure 1: Typical MRI findings in the acute phase of AFM.
Spinal MRIs are shown of an 8-year-old child with AFM, acquired 24 h after onset of neurological symptoms.(A) Sagittal T2 image showing an ill-defined longitudinally extensive central/anterior spinal cord lesion. (B) Axial T2 image from C5–C6 shows hyperintensity of the entire grey matter of the spinal cord, with associated oedema and some surrounding white matter hyperintensity. (C) Axial T2 image from T7 shows asymmetric hyperintensity of the grey matter (right more than left). (D) Axial T2 image from T10 shows hyperintensity of the entire grey matter. (E) Axial FLAIR image at the level of the middle cerebellar peduncle demonstrates hyperintensity of the dorsal pons (arrow). AFM=acute flaccid myelitis.
Acute Management and Differential Treatment Strategies
The acute management of acute flaccid paralysis is largely supportive, focusing on addressing life-threatening complications and providing symptomatic relief, while definitive treatment strategies are guided by the underlying diagnosis. Differential diagnosis directly informs the specific therapeutic approaches employed.
General Supportive Care:
- Respiratory Support: Monitor respiratory function closely, as respiratory muscle weakness is a major complication in AFP, particularly in AFM and GBS. Provide supplemental oxygen, non-invasive ventilation (NIV), or mechanical ventilation as needed. Early intubation should be considered in patients with bulbar weakness or impending respiratory failure.
- Cardiovascular Monitoring: Monitor cardiovascular status, especially in GBS and botulism, where autonomic dysfunction can lead to blood pressure lability and arrhythmias.
- Nutritional Support: Ensure adequate nutrition, considering dysphagia and risk of aspiration. Nasogastric or orogastric feeding tubes or parenteral nutrition may be necessary.
- Bowel and Bladder Management: Manage bowel and bladder dysfunction, which are common in transverse myelitis and spinal cord infarction. Catheterization and bowel regimen may be required.
- Pain Management: Address pain, which can be neuropathic (GBS, AFM), musculoskeletal, or related to immobility. Multimodal pain management strategies may include analgesics, neuropathic pain medications, and physical therapy.
- Prevention of Complications: Prevent complications of immobility, such as pressure ulcers, deep vein thrombosis (DVT), and infections. Regular repositioning, pressure-relieving devices, DVT prophylaxis, and meticulous skin care are essential.
- Rehabilitation: Initiate early rehabilitation, including physical therapy, occupational therapy, and speech therapy, to maintain joint mobility, prevent contractures, and optimize functional recovery.
Diagnosis-Specific Treatments:
- Guillain-Barré Syndrome (GBS):
- Immunomodulatory Therapy: Intravenous immunoglobulin (IVIG) or plasma exchange (PLEX) are effective in accelerating recovery and reducing disability in GBS. These therapies are typically initiated within two weeks of symptom onset.
- Transverse Myelitis (TM):
- High-Dose Corticosteroids: Intravenous methylprednisolone is the first-line treatment for acute TM to reduce inflammation and improve outcomes.
- Plasma Exchange (PLEX): May be considered in steroid-unresponsive TM or in severe cases.
- Immunosuppressants: For relapsing TM or NMOSD/MOGAD-related TM, long-term immunosuppression with agents like azathioprine, mycophenolate mofetil, rituximab, or eculizumab may be necessary.
- Spinal Cord Infarction:
- Supportive Care: Management is primarily supportive as there is no specific treatment to reverse infarction. Focus on optimizing blood pressure, managing pain, and rehabilitation.
- Thrombolysis or Anticoagulation: In rare cases of suspected thromboembolic etiology within a very narrow time window, thrombolysis or anticoagulation might be considered, but evidence is limited and risks must be carefully weighed.
- Botulism:
- Botulinum Antitoxin: Administer botulinum antitoxin as early as possible to neutralize circulating toxin. Heptavalent botulinum antitoxin is available for most types of botulism.
- Supportive Care: Intensive supportive care, including ventilatory support, is critical due to prolonged paralysis.
- Poliomyelitis:
- Supportive Care: Management is supportive, as there is no antiviral treatment for poliovirus. Focus on respiratory support, preventing complications, and long-term rehabilitation.
- Vaccination: Vaccination is the primary prevention strategy for poliomyelitis.
- West Nile Virus Myelitis, Japanese Encephalitis, Tick-borne Encephalitis:
- Supportive Care: Management is primarily supportive, focusing on managing symptoms and complications.
- Antiviral Therapy: No specific antiviral therapy is established for these arboviral infections, although research is ongoing.
- Periodic Paralysis:
- Potassium Management: For hypokalemic periodic paralysis, administer potassium supplementation during attacks. For hyperkalemic periodic paralysis, administer glucose and insulin or diuretics to lower potassium levels.
- Preventative Measures: Dietary modifications (avoiding high-carbohydrate meals, potassium-rich foods), acetazolamide (for hypokalemic PP), or thiazide diuretics (for hyperkalemic PP) may be used to prevent attacks.
- Acute Flaccid Myelitis (AFM):
- Supportive Care: Management is primarily supportive, as there is no proven effective antiviral or immunomodulatory therapy for AFM.
- Intravenous Immunoglobulin (IVIG): Sometimes used empirically for potential antiviral and immunomodulatory effects, although evidence of benefit is limited.
- Corticosteroids: Generally not recommended and may be harmful in viral myelitis, but may be considered in cases with significant spinal cord edema causing compression.
- Experimental Therapies: Antiviral agents and immunomodulatory therapies are under investigation for AFM, but currently not standard of care.
The differential diagnosis of AFP is paramount in guiding treatment strategies. While supportive care forms the cornerstone of management for all causes of AFP, specific therapies are available for certain conditions, such as immunomodulation for GBS and TM, antitoxin for botulism, and potassium management for periodic paralysis. For AFM and many viral myelitides, supportive care remains the mainstay, highlighting the urgent need for further research into targeted therapies.
Prognosis and Long-Term Considerations in AFP: Implications for Differential Diagnosis
The prognosis and long-term outcomes of acute flaccid paralysis vary significantly depending on the underlying etiology. Accurate differential diagnosis not only guides acute management but also provides crucial information for prognostication and long-term care planning.
Prognosis Variability Based on Etiology:
- Acute Flaccid Myelitis (AFM): Prognosis for motor recovery in AFM is variable. Many patients experience persistent weakness and disability, particularly in the affected limbs. Recovery is often incomplete, with residual muscle atrophy and functional impairment. Cranial nerve deficits may have better recovery potential than limb weakness. Long-term sequelae include musculoskeletal deformities, limb length discrepancies, scoliosis, and neurological deficits.
- Guillain-Barré Syndrome (GBS): Prognosis is generally favorable with immunomodulatory treatment. Most patients achieve significant recovery, although some may have residual weakness, fatigue, and sensory symptoms. Mortality is low with modern supportive care, but severe cases with autonomic dysfunction or respiratory failure carry a higher risk.
- Transverse Myelitis (TM): Prognosis is variable depending on the etiology and severity. Recovery ranges from minimal to substantial, with many patients experiencing residual weakness, spasticity, sensory deficits, and bowel/bladder dysfunction. NMOSD and MOGAD-related TM may have a relapsing course, requiring long-term immunosuppression.
- Spinal Cord Infarction: Prognosis is often poor, with significant residual paralysis and disability. Recovery is limited, and patients often require long-term rehabilitation and assistive devices.
- Poliomyelitis: Prognosis for motor recovery is variable. Paralysis is often permanent, and patients may develop post-polio syndrome decades later, characterized by new muscle weakness and fatigue.
- Botulism: Prognosis is generally good with antitoxin and supportive care. Recovery can be slow, taking weeks to months, due to the need for nerve terminal regeneration. Long-term sequelae are rare, but fatigue may persist.
- Periodic Paralysis: Prognosis is good with appropriate management of potassium levels and preventative strategies. Attacks are episodic, and patients can lead relatively normal lives between attacks.
Long-Term Management and Rehabilitation:
Regardless of the specific diagnosis, long-term management of AFP focuses on maximizing functional recovery, minimizing disability, and addressing long-term sequelae. Key aspects include:
- Comprehensive Rehabilitation: Continued physical therapy, occupational therapy, and speech therapy are crucial for optimizing motor function, mobility, activities of daily living, and communication.
- Orthotics and Assistive Devices: Orthoses, braces, wheelchairs, and other assistive devices may be necessary to improve mobility and function.
- Pain Management: Chronic pain management strategies may be needed for neuropathic pain, musculoskeletal pain, or spasticity-related pain.
- Management of Spasticity: Spasticity, if present (e.g., in TM, spinal cord infarction), can be managed with medications, physical therapy, and botulinum toxin injections.
- Bowel and Bladder Care: Long-term management of bowel and bladder dysfunction may involve medications, intermittent catheterization, and bowel management programs.
- Psychosocial Support: Psychological support and counseling are essential to address the emotional and social impact of chronic disability on patients and their families.
- Surgical Interventions: In select cases of AFM and polio, nerve transfer surgery, tendon transfer surgery, or orthopedic procedures may be considered to improve function in severely affected limbs.
- Long-Term Follow-up: Regular neurological and rehabilitation follow-up is necessary to monitor for disease progression, manage complications, and adjust treatment plans as needed.
Prognostic Indicators and Differential Diagnosis:
Certain clinical and investigative findings can help predict prognosis in AFP, and these are often linked to the differential diagnosis.
- Severity of Initial Weakness: More severe initial weakness, particularly complete paralysis, is generally associated with a poorer prognosis for motor recovery across various etiologies.
- Rate of Progression: Rapidly progressive AFP, as seen in spinal cord infarction, may indicate a less favorable prognosis compared to more gradually progressive conditions like GBS with timely treatment.
- MRI Findings: The extent and location of spinal cord lesions on MRI can correlate with prognosis in AFM and TM. More extensive gray matter involvement in AFM and severe white matter damage in TM may predict poorer outcomes. In spinal cord infarction, the extent of infarction on MRI is a prognostic factor.
- Electrophysiological Findings: Severely reduced or absent CMAPs on EMG/NCS in AFM and GBS indicate more severe axonal injury and may predict poorer motor recovery.
- CSF Findings: CSF findings are more useful for differential diagnosis than for prognostication, but persistent inflammation in CSF may indicate ongoing disease activity in TM.
- Response to Acute Treatment: Response to immunomodulatory therapy in GBS and TM is a positive prognostic indicator. Lack of response or delayed response may suggest a poorer outcome or need for alternative therapies.
Understanding the prognostic implications of different AFP etiologies is crucial for counseling patients and families, setting realistic rehabilitation goals, and planning long-term care. Differential diagnosis not only guides acute treatment but also shapes expectations for recovery and long-term management strategies.
Conclusion: Mastering the Differential Diagnosis of Acute Flaccid Paralysis for Improved Patient Outcomes
Acute flaccid paralysis represents a complex and challenging clinical syndrome demanding a systematic and comprehensive approach to differential diagnosis. The diverse etiologies, ranging from infectious diseases like AFM and poliomyelitis to immune-mediated conditions like GBS and transverse myelitis, and structural causes such as spinal cord infarction, underscore the critical need for accurate and timely diagnosis.
Mastering the differential diagnosis of AFP relies on a thorough integration of clinical assessment, epidemiological context, neuroimaging, CSF analysis, electrophysiological studies, and targeted laboratory investigations. Recognizing subtle differences in clinical presentation – such as the pattern of weakness, sensory involvement, reflexes, associated symptoms, and temporal profile – is the cornerstone of initial differentiation. Neuroimaging, particularly spinal MRI, plays a pivotal role in visualizing spinal cord lesions and distinguishing between gray matter predominant involvement in AFM, white matter lesions in TM, and infarction patterns. CSF analysis provides crucial information about inflammation and infection, while EMG/NCS helps differentiate between neuropathic, myopathic, and neuromuscular junction disorders. Targeted laboratory tests, including viral PCR, serology, and antibody assays, are essential for confirming specific etiologies.
The diagnostic journey for AFP is not merely an academic exercise; it has profound implications for patient care. Accurate differential diagnosis directly guides acute management strategies, determining the use of immunomodulatory therapies, antitoxins, antiviral agents, or potassium management, in addition to essential supportive care. Furthermore, understanding the underlying etiology is crucial for prognostication, allowing clinicians to counsel patients and families realistically about expected recovery trajectories and potential long-term sequelae. It informs the planning of comprehensive rehabilitation programs and the anticipation of long-term medical and psychosocial needs.
Continued research is essential to refine diagnostic criteria, develop more sensitive and specific diagnostic tests, and explore targeted therapies for conditions like AFM and viral myelitides, where treatment options are currently limited. Enhancing clinician education and awareness regarding the nuances of AFP and its differential diagnosis is paramount to improve patient outcomes globally. By mastering the art and science of AFP differential diagnosis, healthcare professionals can significantly impact the lives of individuals affected by this devastating syndrome, ensuring timely, appropriate, and personalized care that optimizes recovery and minimizes long-term disability.
Figure 2: Diagnostic criteria for AFM.
These criteria apply to the acute stage of the disease. AFM=acute flaccid myelitis. H=history. E=examination. CSF=cerebrospinal fluid. P=diagnostic element is present. A=diagnostic item is absent. P/A=presence of this diagnostic element is supportive but not required. ND=test was not done. ADEM=acute disseminated encephalomyelitis. MOG=myelin oligodendrocyte glycoprotein. *Subjective (H1) or objective (E1) weakness must be present in any of: limb(s), neck, or cranial nerves. †Prodromal illness can include respiratory, gastrointestinal, or other symptoms of viral illness. ‡Normal or increased reflexes can be found in other limbs. §If MRI obtained very early (within hours of neurological onset) appears normal, repeat MRI after clinical evolution might show diagnostic findings. MRI obtained at late stages (≥4 weeks) might be normal. ¶CSF may be normal at very early (hours) or late (≥4 weeks) stages of AFM. ||At present, there are no data describing the frequency of these features in patients with AFM.