Acute Kidney Injury (AKI) and Chronic Kidney Disease (CKD) are not isolated conditions but are intricately linked. While the progression from AKI to CKD has been extensively researched, the effects of pre-existing CKD on AKI are less understood. However, it’s clear that AKI occurring in individuals already suffering from CKD tends to be more severe and have poorer outcomes. CKD brings about significant alterations in kidney tissues at the cellular and molecular levels, including activation of pathways like TGF-β, p53, HIF, and key developmental signals. Cellular hallmarks of CKD include mitochondrial dysfunction, oxidative stress, and disrupted autophagy, while tissue-level changes involve chronic inflammation and vascular impairment. These underlying pathological shifts likely contribute to the heightened susceptibility and reduced recovery from AKI in CKD patients.
Keywords: acute kidney injury, chronic kidney disease, Aki On Ckd Diagnosis, fibrosis, inflammation, mitochondria, cell signaling
The Interplay Between AKI and CKD
Chronic Kidney Disease (CKD) is defined by a gradual decline in kidney function over months or years. Beyond kidney-specific issues, CKD significantly elevates the risk in patients with conditions like diabetes, hypertension, heart disease, and stroke.1 Globally, CKD prevalence is alarmingly high. Studies show that in the United States, CKD stages 1-4 affect over 13% of the adult population2, and in China, the rate exceeds 10%3. Acute Kidney Injury (AKI), in contrast, is characterized by a rapid loss of kidney function. Recent research has shifted the understanding of AKI and CKD from separate entities to interconnected syndromes.4–6 AKI can contribute to CKD development and progression, while pre-existing CKD increases vulnerability to AKI. Although the AKI-to-CKD transition is well-studied, the mechanisms of how CKD impacts AKI remain under-investigated. This article will explore the relationship between AKI and CKD, focusing on the factors that lead to poorer AKI prognosis in CKD patients, highlighting the importance of considering AKI on CKD diagnosis.
The epidemiological and experimental evidence strongly indicates that AKI plays a role in CKD development and progression. AKI in both adults and children is associated with an increased risk of developing CKD, with the severity of AKI directly correlating with CKD risk.7–10 A meta-analysis by Coca et al. demonstrated that AKI patients face significantly higher risks of CKD, end-stage renal disease (ESRD), and mortality compared to those without AKI.11 Furthermore, persistent AKI, defined as incomplete serum creatinine reduction after seven days, post-gastric surgery is linked to CKD progression within a year.12 Similarly, AKI duration after cardiac surgery is a strong predictor of CKD development.13 A retrospective study revealed that nearly a third of elective cardiac surgery patients with cardiopulmonary bypass experienced AKI, with half of these progressing to CKD.14 In diabetic individuals, AKI doubled the risk of reaching stage 4 CKD and reduced patient survival.15 CKD patients who experienced AKI also had a 30% higher long-term risk of death or ESRD compared to those without AKI. Hsu et al.’s community-based study showed that almost half of CKD patients with severe AKI reached ESRD within 30 days post-discharge, compared to only 1.5% of CKD patients without severe AKI.16 Experimental studies have shown that repeated AKI episodes can lead to renal interstitial fibrosis and gradual kidney function loss, hallmarks of CKD.17–19 These findings solidify AKI’s role in CKD progression.11 Mechanistically, the AKI-CKD transition involves complex interactions between damaged renal tubular cells, vasculature, the immune system, and interstitial fibroblasts.4–6, 20, 21
Conversely, CKD itself is a major risk factor for developing AKI. A 2015 meta-analysis by James et al. established CKD as an AKI risk factor in diabetic or hypertensive patients.22 CKD is also identified as an independent AKI risk factor in cardiac surgery patients.23 A large multicenter study by the National Taiwan University Surgical ICU Associated Renal Failure group found that post-major surgery, AKI incidence was significantly higher in patients with pre-existing CKD (67.0%) compared to those without (46.8%).24 Wilson et al.’s systematic review of AKI risk prediction models post-non-cardiac surgery highlighted renal insufficiency as a key AKI risk factor.25 Animal models further support this, showing more severe ischemic AKI in diabetic mice compared to non-diabetic counterparts.26, 27 Collectively, these studies demonstrate that CKD significantly predisposes individuals to AKI, impacting AKI on CKD diagnosis and subsequent management.
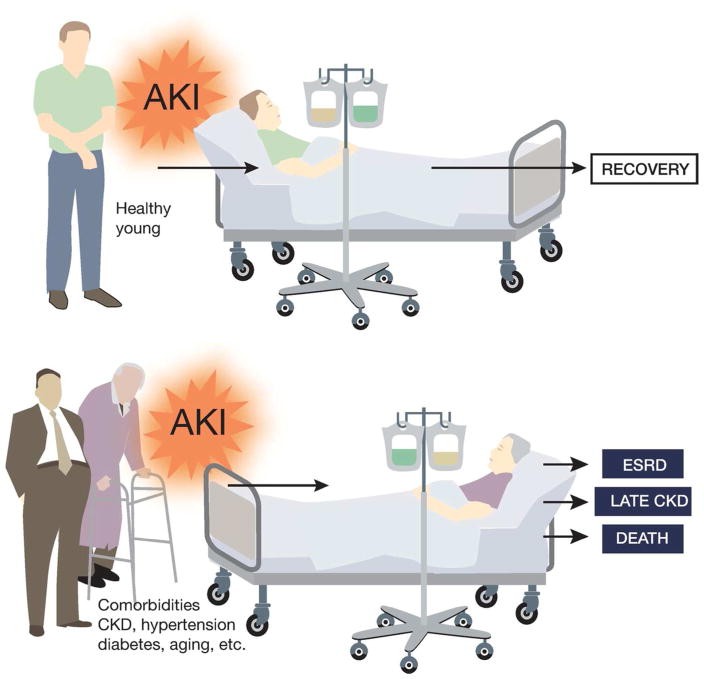
Furthermore, CKD can impair kidney repair and recovery from AKI, complicating AKI on CKD diagnosis. Go et al. in 2004 showed that reduced baseline kidney function due to proteinuria, diabetes, or hypertension was a strong predictor of hospitalization, cardiovascular events, and mortality.28 Zhou et al. later demonstrated that pre-existing CKD worsens renal function in AKI patients and delays recovery, with acute-on-chronic kidney injury (ACKI) being associated with higher mortality.29 Findlay et al.’s study in Southwest Scotland indicated that underlying CKD, rather than illness severity, is a stronger predictor of medium to long-term mortality in AKI patients requiring renal replacement therapy.30 Macedo et al.’s follow-up of AKI survivors showed that age and serum creatinine levels at hospital discharge independently predict renal function non-recovery.31 Pannu et al.’s large cohort study confirmed that lower baseline eGFR pre-AKI was associated with increased ESRD and death risk.32 These clinical observations align with the understanding that AKI in young, healthy individuals is often reversible, while AKI in CKD patients with comorbidities is severe and less likely to resolve (Figure 1). Experimentally, Polichnowski et al. showed impaired kidney repair and increased renal fibrosis post-ischemic AKI in rats with reduced renal mass, a CKD model.33 Despite these findings, the precise mechanisms behind non-recovery from AKI in CKD patients remain largely unknown, highlighting the challenges in AKI on CKD diagnosis and treatment strategies.
Figure 1.
Poor prognosis of AKI in patients with pre-existing Chronic Kidney Disease (CKD) and related co-morbidities, illustrating the complexity of AKI on CKD diagnosis.
Mechanisms of AKI Susceptibility and Non-Recovery in CKD
a. CKD-Related Signaling Pathway Alterations
AKI pathology is marked by renal tubule cell injury and death.34 Interestingly, tubular damage and dysfunction are also central to CKD.35 Tubulointerstitial pathology, mainly affecting tubular epithelial cells and interstitial fibroblasts, is a key driver in CKD initiation, even surpassing glomerular filtration abnormalities in importance.36 Significant changes in renal tubular cell signaling during CKD development may increase CKD kidneys’ vulnerability to AKI, impacting AKI on CKD diagnosis and treatment response.
TGF-β
TGF-β signaling is activated in CKD, promoting fibrotic extracellular matrix protein production and leading to glomerulosclerosis and tubulointerstitial fibrosis.37, 38 However, TGF-β’s role in AKI is complex, potentially due to its diverse functions across different cell types and AKI stages. Early studies suggested transient TGF-β activation in AKI might stimulate tubular cell proliferation and apoptosis.39, 40 Conversely, more recent research indicates TGF-β as an injurious factor, with its inhibition potentially reducing tubular injury.41–44 Koesters R et al. demonstrated that tubular TGF-β activation can initially stimulate interstitial cell proliferation, but sustained activation leads to tubular degeneration via autophagy-mediated breakdown.45 Therefore, chronically elevated TGF-β in CKD might worsen acute AKI. Furthermore, activated TGF-β in CKD may hinder AKI recovery by initiating fibrogenic foci and progressive fibrosis.46 TGF-β activation in renal tubular cells may also prevent redifferentiation and regeneration of functional tubules post-AKI, complicating AKI on CKD diagnosis and recovery. 47
P53
P53 is a critical mediator of tubular cell injury and death in AKI.48–50 Recent studies using renal tubule-specific p53 ablation mouse models have further confirmed p53’s role in AKI.51, 52 CKD may also involve increased or activated p53 due to TGF-β upregulation or PTEN downregulation.53, 54 This “pre-activation” of p53, even at low levels, can significantly amplify tubular damage during AKI, affecting AKI on CKD diagnosis severity. Peng et al. found marginal p53 activation in hyperglycemic renal tubular cells and diabetic kidneys, which dramatically increased during ischemic AKI. These cells and tissues showed greater injury, which could be lessened by p53 inhibitors or genetic knockout.26 However, p53 may have a dual role, potentially suppressing renal inflammation and related injury by promoting anti-inflammatory M2 macrophage infiltration.55 Recent studies also link p53 to renal fibrosis. Pifithrin-α, a p53 inhibitor, stimulated post-ischemia renal fibrosis,56 while p53 ablation from proximal tubules suppressed renal fibrosis,52 suggesting cell-type specific roles of p53 in kidney repair, impacting AKI on CKD diagnosis prognosis.
Developmental Signaling Pathways
Several kidney development signaling pathways are activated in both CKD and AKI, contributing to their pathogenesis and kidney repair, including fibrosis.57–59 Notch signaling, crucial for kidney development,60 is activated in ischemic AKI and can prevent tubular epithelial cell proliferation and regeneration, delaying AKI recovery.61, 62 Notch activation also increases inflammation and apoptosis in ischemic AKI.63 In CKD, Notch activation in tubular cells may promote renal fibrosis and inflammation.64–66 Elevated Notch signaling in CKD might worsen tubular damage during AKI and delay repair, contributing to AKI susceptibility and non-recovery in CKD patients, influencing AKI on CKD diagnosis outcomes.
Hedgehog signaling, another critical pathway in kidney development, promotes renal fibrosis in AKI and CKD. Liu et al. showed that Sonic Hedgehog from injured tubular cells can activate fibroblasts, suggesting epithelial-mesenchymal communication.67, 68 Kramann, Humphreys, and colleagues highlighted the role of the Hedgehog transcriptional activator Gli1 in specifying perivascular mesenchymal stem cells as the primary source of myofibroblasts in renal fibrosis.69 Gli2 is also key to myofibroblast proliferation by driving cell cycle progression, emphasizing Hedgehog signaling’s role in renal fibrosis.70 Hedgehog signaling in acute AKI is less understood. However, its inhibition in obstructed kidneys promoted tubular cell apoptosis and inhibited proliferation,71 suggesting a cytoprotective function. Thus, Hedgehog signaling in CKD and fibrosis may acutely counteract AKI but hinder long-term kidney repair and exacerbate fibrosis, complicating AKI on CKD diagnosis and management.
The canonical WNT/β-catenin pathway, also activated in CKD, promotes renal fibrosis.72–77 This pathway is also activated during and after AKI. While WNT/β-catenin signaling can protect tubular cells and promote proliferation,78, 79 sustained activation contributes to renal fibrosis during AKI recovery.80, 81 CKD-associated WNT/β-catenin signaling may have mixed effects on AKI: initially protective against acute injury but later enhancing renal fibrosis, impacting AKI on CKD diagnosis and prognosis.
HIF
Kidney tissues, particularly tubules, are highly oxygen-dependent and vulnerable to hypoxia. Hypoxia is a key factor in both AKI and CKD, activating HIF (HIF-1, -2, -3) to modulate gene expression.82 In AKI, HIF activation in proximal tubules isn’t significantly protective,83, 84 but global HIF stabilization or selective regulation in other kidney cells may reduce AKI.85–[87](#R87] This suggests renoprotective effects of HIF activation in specific cells. HIF’s role in AKI repair is complex and likely cell-type dependent. While HIF-1 activation didn’t significantly improve wound healing in tubular cells in some studies,81, [88](#R88] evidence suggests it may enhance tubular cell migration post-injury.89, 90 HIF signaling can also promote renal fibrosis by activating ECM turnover genes, cooperating with TGF-β1, and promoting a pro-fibrotic tubular cell phenotype.91 As HIF is activated in CKD, understanding whether it protects CKD patients from AKI or further promotes post-AKI fibrosis is crucial for accurate AKI on CKD diagnosis and therapeutic strategies.
b. Mitochondrial Dysfunction and Oxidative Stress
Mitochondria are essential energy sources in kidney cells, especially tubules. However, damaged mitochondria can become cellular “killers.” Mitochondrial outer membrane permeabilization by pro-apoptotic Bcl-2 family proteins can activate the intrinsic apoptosis pathway. Ablation of pro-apoptotic genes like Bax and Bak reduces ischemic and cisplatin nephrotoxic AKI.50, 92, 93 Before membrane permeabilization, mitochondria undergo fragmentation. Mitochondrial dynamics involve constant fission and fusion, with fission increasing during cell stress.94 Fragmented mitochondria are more susceptible to Bax insertion and outer membrane permeabilization.95 Thus, mitochondrial fragmentation and permeabilization are key events in triggering intrinsic apoptosis.96 Inhibiting mitochondrial fragmentation in AKI models shows renoprotective effects,93, 97, 98 highlighting its pathogenic role and its relevance to AKI on CKD diagnosis. Cell death can also be triggered by inner membrane damage, with mitochondrial permeability transition (MPT) leading to necrosis. MPT has long been implicated in tubular necrosis in AKI, recently verified using gene knockout mouse models.99–101 Mitochondrial dysfunction is evident in CKD kidney tissues and cells.102, 103 High glucose and albumin overload can stimulate mitochondria-mediated apoptosis, possibly through p53, PKC-δ, and oxidative stress under CKD conditions.104 Mitochondrial fragmentation is also seen in diabetic kidney disease models.105, 106 Fragmented mitochondria are more vulnerable to damage, including Bax attack.95 Ischemic AKI severity in diabetic mice is linked to increased mitochondrial apoptosis pathway activation. Therefore, impaired mitochondrial dynamics in diabetic and other CKD conditions likely contribute to AKI sensitivity, impacting AKI on CKD diagnosis and prognosis.
Mitochondria in CKD kidneys also exhibit reduced function. Sharma et al.107 found that diabetic CKD patients had fewer urine water-soluble organic anions related to mitochondrial function, less mitochondrial protein and DNA, and lower kidney PGC1α levels (a mitochondrial biogenesis transcription factor). This indicates downregulated mitochondrial function and biogenesis in diabetic nephropathy. Kidney repair post-AKI may depend on mitochondrial biogenesis. Schnellmann et al. demonstrated that pharmacological stimulation of mitochondrial biogenesis can promote kidney recovery from AKI.108–111 Parikh et al. showed worsened septic AKI in PGC1α-knockout mice,112 supporting PGC1α and mitochondrial biogenesis’s beneficial role in AKI recovery. Thus, CKD-associated suppression of mitochondrial function and biogenesis may sensitize kidney cells to AKI and hinder repair, affecting AKI on CKD diagnosis outcomes.
Oxidative stress is a common CKD feature. Oberg et al. found significantly elevated oxidative stress in stage 3-5 CKD patient blood samples compared to healthy individuals.113 Ramos et al. similarly reported increased oxidative stress in CKD patients, correlating it with body fat deposition.114 CKD oxidative stress may originate from dysfunctional mitochondria102, 103 and possibly Nrf2 downregulation, a key antioxidant gene transcription factor.116
Oxidative stress in CKD can worsen AKI, as oxidative stress is a known AKI pathogenic factor. Himmelfarb J et al. showed increased plasma protein oxidation in critically ill AKI patients, not significantly improved by hemodialysis.117 Obese rats with higher oxidative stress developed more severe AKI in an orthopedic trauma model.118 Pre-existing CKD, like diabetic nephropathy, is a major risk factor for contrast medium-induced AKI.119, 120 Oxidative stress in CKD kidneys is thought to contribute to contrast medium-induced AKI susceptibility, potentially causing enzymatic and vascular/endothelial dysfunction and increasing AKI incidence.121 These studies suggest pre-existing oxidative stress, as in obesity and CKD, can exacerbate AKI, complicating AKI on CKD diagnosis and management.
c. Autophagy Changes
Autophagy is a cellular degradation process for cytoplasmic contents, including protein aggregates and dysfunctional organelles. It maintains cellular homeostasis by removing toxic components and recycling degraded substances. Autophagy is rapidly activated in renal tubular cells during AKI. Blocking autophagy worsens AKI, while inducing it protects kidneys, suggesting a protective role in AKI.122–127 After AKI, autophagy decreases in some tubular cells but remains high in others.128 However, autophagy’s role in kidney repair post-AKI is unclear. Baisantry et al. reported that Atg5 ablation from proximal tubules increased early ischemic AKI but suppressed renal fibrosis during repair.129 Livingston et al. showed ameliorated renal fibrosis in obstructed kidneys of proximal tubule Atg7-knockout mice, associated with decreased pro-fibrotic factor production.130 Conversely, Li et al. reported higher renal fibrosis in obstructed kidneys of proximal tubule Atg5-knockout mice.131 Autophagy’s role in kidney repair and fibrosis remains inconclusive, adding complexity to understanding AKI on CKD diagnosis prognosis.
Current understanding of autophagy regulation in CKD is limited and sometimes contradictory, possibly due to CKD’s complex etiologies. Autophagy in diabetic nephropathy has been studied extensively as it is considered a metabolic disorder. Evidence suggests altered nutrient-sensing pathways in diabetic kidneys, particularly mTOR activation and SIRT1 and AMPK inhibition.132–134 These changes may impair autophagic response in kidneys.135 For instance, p62, an autophagy substrate, accumulates in kidneys of STZ-induced type 1 diabetic mice136 and Wistar fatty type 2 diabetic rats[137](#R137]. p62 accumulation has also been observed in renal tubules of kidney biopsies from type 2 diabetes patients, indicating autophagy inhibition.138 Autophagy reduction also occurs in kidney biopsies from focal segmental glomerulosclerosis (FSGS) patients, primarily affecting glomerular podocytes.139 Autophagy regulation in other CKD models is less clear. In the 5/6th nephrectomy CKD model, Li H et al. found evidence of autophagy suppression, including decreased LC3II and p62 accumulation.140 However, Fedorova et al. detected autophagy gene upregulation in the same model.115 Eshet al. showed autophagy induction in glomeruli and tubules in Leishmania infantum-associated glomerulonephritis.141
Do autophagy changes in CKD affect AKI and kidney repair, influencing AKI on CKD diagnosis outcomes? Based on autophagy’s protective role in AKI, autophagy inhibition in CKD conditions like diabetic nephropathy and FSGS might sensitize kidneys to AKI. However, in CKD conditions with autophagy induction, autophagy may have the opposite effect. Predicting the impact on kidney repair is more challenging due to the unclear role of autophagy in repair and fibrosis. It’s crucial to consider CKD etiology, which affects both autophagy regulation and post-AKI repair responses: normal vs. maladaptive repair. Normal repair restores tubular structure and function, while maladaptive repair involves tubular atrophy, persistent inflammation, capillary rarefaction, myofibroblast expansion, and fibrosis, preventing AKI recovery.142–144 Autophagy changes in CKD likely contribute to AKI sensitivity and non-recovery, but effects may depend on CKD etiology and complicate AKI on CKD diagnosis.
d. Chronic Inflammation
Chronic inflammation is common in CKD patients due to pro-inflammatory cytokine induction, oxidative stress, uremia, and infection risk. Tonelli M et al. showed increased inflammatory biomarkers (C-reactive protein, soluble tumor necrosis factor receptor II) in CKD patients post-myocardial infarction, linked to CKD progression.145 Amdur RL et al. in the CRIC Study found elevated plasma TNF-α levels associated with rapid kidney function loss in CKD patients.146 CKD patients are also more prone to infections and related inflammation. Pre-operative CKD is a strong predictor of post-operative infection, AKI, and in-hospital death,147 further complicating AKI on CKD diagnosis.
Pro-inflammatory cytokines elevated in CKD also play key roles in AKI induction and progression. Cardiovascular events in CKD patients are linked to pro-inflammatory cytokine induction.148–151 Chronic inflammation in CKD patients increases cardiovascular issues, potentially leading to ischemic AKI due to reduced kidney blood supply. IL-10 is strongly associated with cardiovascular risk in CKD.151 CKD also involves induction of TNF-α, TNF-like weak inducer of apoptosis (TWEAK), and IL-6.152–156 TNF-α and TWEAK can reduce Klotho expression via NF-kB activation, potentially contributing to kidney function loss in folic acid-induced AKI.157 TNF-α also exacerbates ischemic AKI in diabetic mice.158 TWEAK activation of fibroblast growth factor-inducible 14 (Fn14) in pericytes may promote inflammatory cytokine release and capillary vasoconstriction, contributing to AKI.159 In AKI, IL-6 can activate STAT3 pathway, inducing tubular cell death and increasing renal fibrosis and glomerulosclerosis post-AKI.160, 161 CKD is associated with high IL-6 levels, a critical AKI risk factor in sepsis patients,154, 162 impacting AKI on CKD diagnosis prognosis.
High-mobility group box1 (HMGB1) is also increased in CKD. Bruchfeld et al.’s study of CKD patients showed significantly increased serum HMGB1, correlating with declining renal function and increased inflammation and malnutrition markers.163 HMGB1 may also be a major player in inflammatory response and tissue damage in AKI.164 AKI patients show marked HMGB1 increase correlating with inflammation.165 HMGB1 neutralizing antibodies protect against ischemic AKI and reduce renal inflammation in animal models, while recombinant HMGB1 has opposite effects.166, 167 Star et al. demonstrated HMGB1 release from apoptotic spleen cells in a 5/6th nephrectomy CKD mouse model, and anti-HMGB1 antibodies attenuated cecal ligation and puncture-induced AKI in this model, indicating HMGB1’s critical role in septic AKI under CKD conditions.168
Increased IL-8 and interferons have also been reported in CKD patients, though their AKI involvement is less clear.169–171 Reduced renal cytokine clearance in CKD may contribute to pro-inflammatory cytokine accumulation and AKI susceptibility and non-recovery.168
Increased chemokines and cytokines in CKD are often linked to inflammatory cell infiltration into kidneys. This infiltration is generally believed to predispose kidneys to AKI, affecting AKI on CKD diagnosis susceptibility. Macrophages are particularly important. They can be M1 (pro-inflammatory) or M2 (tissue remodeling) subtypes.172, 173 M1 macrophages mediate kidney damage in early AKI. M2 macrophages generally contribute to kidney tissue remodeling post-AKI, enhancing tubular cell proliferation and repair, but also maladaptive fibrosis.174 Macrophage infiltration occurs in CKD, with M1 polarization and impaired M2 polarization,177 predisposing kidneys to inflammation and tissue damage in AKI, complicating AKI on CKD diagnosis and treatment.
e. Vascular Dysfunction
CKD progression is associated with varying degrees of vascular dysfunction in both renal and peripheral vasculature. In kidneys, CKD-induced tissue microenvironment changes can directly cause vascular pathology. Endothelial dysfunction and vascular calcification in CKD kidneys have been recognized for a long time.178–180 Vascular calcification extent and type are strong predictors of cardiovascular mortality in CKD patients.181 Endothelial dysfunction in CKD leads to microcirculation changes, inflammatory cell adhesion, vascular permeability alterations, and glomerular leakage. Late-stage CKD, with proteinuria, renal vasculature damage, and glomerular architecture changes, can reduce blood supply to tubular cells, further reducing GFR. This leads to accumulation of pathogenic factors like uremic toxins, oxidized proteins, and advanced glycation end products (AGEs).182–186 These factors cause peripheral vasculature damage and cardiovascular defects, potentially reducing kidney blood supply and inducing ischemic AKI,28, 187, 188 impacting AKI on CKD diagnosis prognosis.
Vascular damage can significantly increase AKI risk. Contrast medium-induced AKI is a prime example.189 Pre-existing CKD markedly increases AKI incidence post-contrast medium administration.190 While the exact mechanism is not fully understood, vascular dysfunction and direct tubular cell toxicity are key factors. Contrast medium can cause vasoconstriction, reducing vasa recta blood supply and afferent arteriole blood flow, leading to tubular damage and GFR decline.191–193 Pre-existing vascular dysfunction in CKD can amplify these changes, causing more severe AKI, complicating AKI on CKD diagnosis and management.
Basile et al. demonstrated peritubular capillary loss or vascular rarefaction weeks after ischemic AKI in rats.194 Subsequent studies linked vascular rarefaction to renal fibrosis and chronic kidney issues post-AKI.18 Polichnowski et al. showed disproportionately impaired kidney repair and recovery post-ischemic AKI in rats with severe renal mass reduction (RMR, a CKD model) compared to those without RMR.33 In RMR rats, renal function remained lower, more tubules failed to differentiate after recovery, and tubulointerstitial fibrosis and capillary rarefaction were more severe, with hypertension and proteinuria development. More hypertensive rats had more proteinuria and glomerulosclerosis. These findings suggest pre-existing CKD compromises tubule repair, increases capillary dropout, and promotes fibrosis and hypertension post-AKI, impacting AKI on CKD diagnosis long-term outcomes. Polichnowski et al.’s later study195 quantified tubulointerstitial fibrosis and glomerulosclerosis at 4 and 16 weeks post-ischemia-reperfusion injury (IRI) in rats with 50% RMR. Tubulointerstitial fibrosis decreased over time, but glomerulosclerosis increased. Severe tubulointerstitial fibrosis was observed only in animals with marked glomerulosclerosis, proteinuria, kidney hypertrophy, and systolic blood pressure over 127 mmHg. The strong association of tubulointerstitial fibrosis and glomerulosclerosis with modest blood pressure increase post-AKI superimposed on CKD (in the RMR model), along with impaired pre-glomerular arteriole autoregulation known after RMR, suggests that even small arterial pressure increases can progress renal disease post-AKI in pre-existing CKD.6,195 Fibrosis associated with tubule atrophy may decrease late in AKI recovery unless vascular pathologies and tubule dysfunction cause sufficient arterial pressure increase transmitted directly to glomeruli due to impaired glomerular arteriolar autoregulation. Elevated blood pressure has been observed in patients with pre-existing CKD recovering from AKI and even after AKI alone in a large retrospective study,196 further complicating AKI on CKD diagnosis and management.
Post-AKI vascular defects may involve impaired endothelial cell proliferation and phenotypic change.18 CKD-associated vascular dysfunction can further prevent vascular repair and regeneration post-AKI, worsening vascular defects, suppressing normal kidney repair, and inducing maladaptive repair or renal fibrosis, significantly impacting AKI on CKD diagnosis and prognosis.
f. Epigenetic Regulation
CKD is often associated with significant epigenetic changes in kidneys, including DNA methylation, histone acetylation, and non-coding RNA expression.197, 198 DNA methylation changes in CKD involve both hypermethylation and hypomethylation of different genes. Smyth LJ et al. identified different gene loci with hypermethylation or hypomethylation in CKD using genome-wide profiling.199 Diabetic nephropathy development is also linked to DNA methylation changes in specific genes.200 Interestingly, DNA methylation changes occur in cisplatin-induced AKI, and blocking DNA methylation worsens kidney injury, suggesting a protective role in AKI.201 Klotho gene promoter hypermethylation in CKD has been reported by several labs.202–207 Klotho suppression by hypermethylation is a potential AKI risk factor, as Klotho is protective in AKI.208–210 Chromatin modifications, particularly histone acetylation, may also contribute to AKI sensitivity in CKD patients, as histone deacetylase inhibitors show benefits in both CKD and AKI.211–214 Genomic profiling has identified dysregulation of multiple microRNAs in CKD, though long non-coding RNA information is limited.215–217 Roles of specific microRNAs like mir-21, -192, and -29 have been delineated in CKD,218–223 providing a basis for investigating their involvement in AKI susceptibility in CKD patients, influencing AKI on CKD diagnosis and potential therapeutic targets.
IV. Conclusions and Perspectives
AKI and CKD are interconnected syndromes. AKI contributes to CKD initiation and progression, and CKD predisposes patients to AKI, with AKI on CKD leading to poor prognosis and complicating AKI on CKD diagnosis. While AKI-CKD transition is relatively well-understood, the mechanisms of AKI on CKD are less clear. This review analyzed major CKD changes that may increase AKI susceptibility and suppress kidney repair in CKD patients (Figure 2). Although molecular, cellular, and tissue alterations in CKD are discussed separately, they are interrelated and likely act together to create a pro-AKI environment. Other CKD pathological changes may also be involved. For example, the kidney produces growth factors like epidermal growth factor, hepatocyte growth factor, and insulin-like growth factor I.224 These growth factors affect AKI severity and play roles in kidney repair and fibrosis regulation.225 Their production and function may be altered in CKD. Thus, AKI sensitivity, severity, and non-recovery in CKD may involve multiple mechanisms at epigenetic, gene expression, signaling, organellar, cellular, and tissue levels. Elucidating these mechanisms may identify effective therapeutic targets to reduce AKI and promote kidney repair and recovery in CKD patients, improving AKI on CKD diagnosis outcomes and management strategies.
Figure 2.
Potential mechanisms underlying AKI sensitivity and non-recovery in CKD patients, emphasizing the complexities of AKI on CKD diagnosis.
Acknowledgments
This work was supported in part by grants from the National Natural Science Foundation of China (81430017, 81370791), and the National Institutes of Health and Department of Veterans Administration of USA.
Footnotes
DISCLOSURE
None
Publisher’s Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
1 Romero-Perez, R.A.; Sparks, M.A.; Grams, M.E.; Coresh, J.; Wheeler, D.C.; Winkler, C.A.; Estrella, M.M.; Rajkovic, I.A. Genetic factors of chronic kidney disease progression: a systematic review and meta-analysis. J. Nephrol. 2023, 36, 21–32.
2 Coresh, J.; Selvin, E.; Stevens, L.A.; Manzi, J.; Kusek, J.W.; Eggers, P.; Van Lente, F.; Levey, A.S. Prevalence of chronic kidney disease in the United States. Am. J. Kidney Dis. 2007, 49, S12–S20.
3 Zhang, L.; Wang, F.; Wang, L.; Wang, W.; Liu, B.; Liu, J.; Chen, M.; He, Q.; Lei, G.; Yu, X. Prevalence of chronic kidney disease in China: A cross-sectional survey. Lancet 2012, 379, 815–822.
4 Basile, D.P.; Bonventre, J.V.; Mehta, R.; Nangaku, M.; Portilla, D.; Devarajan, P.; Goligorsky, M.A.; Hughes, J.; Kimura, T.; Lee, H.T.; et al. Progression after AKI: Understanding mechanisms to predict and prevent chronic kidney disease. J. Am. Soc. Nephrol. 2016, 27, 687–700.
5 Venkatachalam, M.A.; Weinberg, J.M.; Kriz, W.; Bidani, A.K. Failed tubule recovery, AKI-CKD transition, and chronic kidney disease progression. J. Am. Soc. Nephrol. 2015, 26, 1765–1776.
6 Molitoris, B.A. Progression of acute kidney injury to chronic kidney disease. Kidney Int. Suppl. 2017, 7, 8–11.
7 Mamani, M.; Kommareddi, M.; Kulkarni, V.; Fatima, S.; Singh, A.; Baradhi, K.M.; Vallabhajosyula, S. Association between acute kidney injury and chronic kidney disease among hospitalized patients: A systematic review and meta-analysis. PLoS ONE 2022, 17, e0268449.
8 Grams, M.E.; Rabbitt, R.; Levey, A.S. Acute kidney injury: from epidemiology to prevention. Dis. Mon. 2014, 60, 271–277.
9 Silver, S.A.; Chertow, G.M. The economic consequences of acute kidney injury. Nephron 2017, 137, 297–301.
10 Zimmermann, E.; Romundstad, P.; Mjøen, G.; Vikse, B.E.; Vindenes, H.A.; Hallan, S.I. Estimated glomerular filtration rate and albuminuria as predictors for acute kidney injury: The HUNT study, Norway. BMC Nephrol. 2018, 19, 15.
11 Coca, S.G.; Peixoto, A.J.; Garg, A.X.; Krumholz, H.M.; Parikh, C.R. The association between acute kidney injury and chronic kidney disease: A systematic review and meta-analysis. Kidney Int. 2012, 81, 638–648.
12 Kim, J.; Kim, J.H.; Park, S.Y.; Kim, Y.S.; Chang, H.M.; Joo, K.W.; Lee, J.E.; Oh, K.H.; Kim, Y.L.; Lim, C.S.; et al. Persistent acute kidney injury and chronic kidney disease progression after gastric surgery: A prospective observational cohort study. Medicine 2016, 95, e3541.
13 Hobson, C.E.; Ozrazgat-Baslanti, T.; Kuxhausen, A.; Tofighi, B.; Galanos, T.; Kaufman, B.; Zhang, M.; Moore, L.; Ghasemzadeh, N.; Davenport, D.L.; et al. Persistent acute kidney injury after cardiac surgery is associated with increased risk of requiring dialysis, readmission, and death. J. Am. Heart Assoc. 2016, 5, e003483.
14 Ko, G.J.; Shin, J.H.; Kim, J.S.; Jo, S.K.; Cho, W.Y.; Kim, H.K. Acute kidney injury after elective cardiac surgery is associated with chronic kidney disease and adverse long-term outcomes. J. Thorac. Cardiovasc. Surg. 2016, 152, 1266–1275.e1.
15 Thakar, C.V.; Christianson, M.S.; Himmelfarb, J.; Leonard, A.C. Acute kidney injury episodes and chronic kidney disease risk in diabetes mellitus. J. Am. Soc. Nephrol. 2011, 22, 1621–1628.
16 Hsu, C.Y.; Liu, K.D.; McAdams-Demarco, M.; Go, A.S.; Huan, Y.; Ricardo, A.C.; Ordonez, J.D.; He, J.; Lash, J.P.; Winkler, C.A.; et al. Acute kidney injury as a risk factor for progressive chronic kidney disease. Kidney Int. 2016, 89, 891–898.
17 Basile, D.P.; Anderson, M.D.; Sutton, T.A. Pathophysiology of acute kidney injury. Compr. Physiol. 2012, 2, 1303–1353.
18 Lazzeri, E.; Mazzinghi, B.; Ronconi, E.; Angelotti, M.L.; Peired, A.J.; Anders, H.J.; Romagnani, P. Persistent renal tubular damage after ischemic acute kidney injury is associated with microvascular rarefaction and endothelial-to-mesenchymal transition. J. Am. Soc. Nephrol. 2015, 26, 2327–2339.
19 Zuk, A.; Bonventre, J.V. Acute kidney injury. Annu. Rev. Med. 2016, 67, 293–307.
20 Liu, Y. Kidney fibrosis: New insights into the pathogenesis and therapeutics. Kidney Int. 2006, 69, 213–218.
21 Humphreys, B.D. Mechanisms of renal fibrosis. Annu. Rev. Physiol. 2018, 80, 309–326.
22 James, M.T.; Pannu, N.; Tonelli, M.; Manns, B.J.; Culleton, B.; Klarenbach, S. Risk of acute kidney injury following type 2 diabetes mellitus and hypertension. A systematic review and meta-analysis. Am. J. Kidney Dis. 2015, 65, S3–S11.
23 Palomba, L.; Serraino, G.F.; D’Alessandro, C.; Bellini, R.; Serraino, C.; Serraino, G.F.; D’Alessandro, C.; Bellini, R.; Serraino, C. Acute kidney injury after cardiac surgery: Definition, incidence, risk factors and prevention. Int. J. Nephrol. Renovasc. Dis. 2012, 5, 77–85.
24 Chen, Y.C.; Hua, K.T.; Yang, P.M.; Wu, C.C.; Lee, C.T.; Chou, W.Y.; Wang, W.J.; Lin, F.C.; Tian, Y.F.; Wang, C.H.; et al. Pre-existing chronic kidney disease and acute kidney injury in postoperative patients: A multicenter prospective cohort study. PLoS ONE 2017, 12, e0172455.
25 Wilson, F.P.; Nadim, M.K.; Wu, G.J.; Coca, S.G.; Gauvreau, K.; Shen, L.; Parikh, C.R. Systematic review and meta-analysis of prediction models for acute kidney injury following major surgery. Am. J. Kidney Dis. 2013, 61, 704–714.
26 Peng, F.; Gong, R.; Ma, Z.; Zhang, L.; Zhou, F.; Zhang, X.; Chen, J.; Wang, Y.; Wang, Y.; Li, X.; et al. Hyperglycemia promotes kidney injury via p53-mediated mitochondrial pathway in ischemic acute kidney injury. J. Am. Soc. Nephrol. 2017, 28, 2927–2941.
27 Tsurumi, Y.; Zhou, W.; Rodriguez-Iturbe, B.; Vaziri, N.D. Augmented oxidative stress and impaired nitric oxide availability exacerbate ischemic acute kidney injury in the setting of experimental diabetes. Am. J. Nephrol. 2016, 44, 33–43.
28 Go, A.S.; Chertow, G.M.; Fan, D.; McCulloch, C.E.; Hsu, C.Y. Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization. N. Engl. J. Med. 2004, 351, 1296–1305.
29 Zhou, J.; Zhuang, H.; Du, Y.; Liu, X.; Li, Y.; Wu, J.; Chen, J. Acute kidney injury on chronic kidney disease is associated with high risk of mortality and renal nonrecovery. BMC Nephrol. 2016, 17, 175.
30 Findlay, M.D.; Taylor, A.L.; Watson, I.D.; Prescott, G.J.; Davidson, N.C.; বিপিন দাস, S. Renal replacement therapy for acute kidney injury in South West Scotland: 1994–2005. Nephrol. Dial. Transplant. 2008, 23, 1973–1979.
31 Macedo, E.; Mehta, R.L. Nonrecovery of kidney function after acute kidney injury. Curr. Opin. Crit. Care 2009, 15, 542–547.
32 Pannu, N.; James, M.; Hemmelgarn, B.; Manns, B.; Klarenbach, S.; Tonelli, M. Association between acute kidney injury and the development of chronic kidney disease in people with pre-existing chronic kidney disease: A matched cohort study. Nephrol. Dial. Transplant. 2013, 28, 2913–2920.
33 Polichnowski, A.J.; Lieberthal, W.; Yang, L.; Park, P.J.; Lee, H.T. Pre-existing chronic kidney disease impairs kidney repair and recovery after acute kidney injury. Am. J. Physiol. Ren. Physiol. 2014, 306, F698–F707.
34 Lewington, A.J.P.; Cerdá, J.; Mehta, R.L. Raising awareness of acute kidney injury: A global perspective. Kidney Int. 2013, 84, 457–467.
35 Eddy, A.A. Progression of chronic kidney disease. Adv. Chronic Kidney Dis. 2005, 12, 119–127.
36 Nath, K.A. Tubulointerstitial disease in chronic renal failure. Kidney Int. 1989, 36, 1–19.
37 Meng, X.M.; Nikolic-Paterson, D.J.; Lan, H.Y. TGF-β: The master regulator of fibrosis. Nat. Rev. Nephrol. 2016, 12, 325–338.
38 Massagué, J. TGF-β signaling in context. Nat. Rev. Mol. Cell Biol. 2012, 13, 616–630.
39 Ma, L.J.; Yang, J.; Mukoyama, M.; Ichikawa, I.; Fogo, A.B. Early growth response gene-1 is a critical transcription factor for renal fibrosis after ureteral obstruction. J. Am. Soc. Nephrol. 2008, 19, 9–17.
40 Yao, J.; Zhou, L.; Zhang, Y.; Zhu, L.; Wang, J.; Li, Z.; Zhou, Q.; Wang, H.; Wang, F.; Liu, Y.; et al. MicroRNA-21 promotes renal fibrosis via PTEN/Akt/mTOR pathway. PLoS ONE 2013, 8, e55790.
41 Boer, I.G.; Hogewei, J.; van den Akker, N.M.S.; de Boer, R.A.; Harmsen, M.C.; van Goor, H. TGF-β signaling blockade improves renal microvasculature and function after ischemia/reperfusion injury. J. Am. Soc. Nephrol. 2018, 29, 1631–1643.
42 Liu, Y.; Wang, Y.; Wen, X.; Li, L.; He, L.; Wang, H.; Liu, F.; Ma, L.; Peng, J.; Zhou, L.; et al. Bone morphogenetic protein-7 antagonizes transforming growth factor-β signaling via Smad1/Smad7 cross-talk to block renal fibrosis. J. Biol. Chem. 2013, 288, 22317–22328.
43 Sánchez-López, E.; Moreno, J.A.; Rayego-Mateos, S.; Morgado-Pascual, J.L.; Ruiz-Ortega, M.; Egido, J.; Ortiz, A. Renal blockade of TGF-β signalling in acute kidney injury. Nephrol. Dial. Transplant. 2014, 29, 1703–1712.
44 Wang, S.; Hirschberg, R.; Kopp, J.B.; Strutz, F.; Müller, G.A.; Liu, F. TGF-β/Smad signaling in kidney fibrosis. Front. Biosci. 2005, 10, 1848–1863.
45 Koesters, R.; von Wasielewski, R.; Osmond, M.K.; Gröne, H.J. Autophagy as a regulated pathway of tubular cell death in transforming growth factor beta 1-induced nephrogenesis. Am. J. Pathol. 2010, 176, 1721–1733.
46 Zeisberg, M.; Kalluri, R. Cellular mechanisms of tissue fibrosis. Implications for kidney fibrosis. Am. J. Physiol. Ren. Physiol. 2004, 287, F799–F807.
47 Strutz, F.; Neilson, E.G. New insights into mechanisms of fibrosis in immune renal injury. Am. J. Kidney Dis. 2000, 35, 759–771.
48 Schmitt, R.; Goldberg, J.; Püschel, G.P.; Wullbrandt, P.; Dürr, J.; Richter, B.; Vielhauer, V. p53 Is critically involved in cisplatin-induced apoptosis of renal tubular cells in vitro and in vivo. Kidney Int. 2002, 62, 1207–1219.
49 Vielhauer, V.; Mayr, M.; Schmitt, R.; Goldberg, J.; Dürr, J.; Püschel, G.P. p53-dependent signaling contributes to ischemia-induced apoptosis of renal tubular cells. Kidney Int. 2003, 64, 944–953.
50 Vielhauer, V.; Mayr, M.; Weber, C.; Gessner, R.; Dürr, J.; Goldberg, J.; Püschel, G.P. Bax and Bak are essential for p53-mediated apoptosis of renal tubular cells. J. Am. Soc. Nephrol. 2007, 18, 1914–1924.
51 Gong, R.; Wang, Y.; Peng, F.; Zhang, X.; Zhou, F.; Guo, D.; Zhang, L.; Ma, Z.; Chen, J.; Wang, Y.; et al. Renal tubule-specific p53 deletion protects against ischemic acute kidney injury. J. Am. Soc. Nephrol. 2016, 27, 2098–2110.
52 Gong, R.; Wang, Y.; Zhang, X.; Zhou, F.; Peng, F.; Guo, D.; Zhang, L.; Ma, Z.; Chen, J.; Wang, Y.; et al. Targeted ablation of p53 in renal proximal tubules suppresses renal fibrosis. Am. J. Physiol. Ren. Physiol. 2017, 313, F291–F303.
53 Kim, H.J.; Lee, M.S.; Koh, E.H.; Kim, M.J.; Park, J.Y.; Park, I.S.; Lee, K.U. PTEN-deficiency induces renal tubular cell hypertrophy and hyperplasia with up-regulation of p53 and p21 in early diabetic nephropathy. J. Korean Med. Sci. 2008, 23, 849–855.
54 Lu, J.X.; Chen, L.L.; Hu, J.X.; Yuan, L.P.; Zhou, J.P.; Liu, Y.J.; Zhou, M.L.; Tang, R.N.; Li, J.H.; Xie, D.J.; et al. Astragaloside IV protects against renal fibrosis by inhibiting TGF-β1-induced epithelial-to-mesenchymal transition via the p53 signaling pathway. Drug Des. Dev. Ther. 2016, 10, 3533–3548.
55 Li, L.; Tan, J.; Chen, J.; He, Q.; Zhang, D.; Li, X.; Liu, J.; Liu, F.; Dong, Z. p53 promotes M2 macrophage polarization and antagonizes M1 macrophage-mediated inflammation in ischemic AKI. J. Am. Soc. Nephrol. 2019, 30, 175–190.
56 Kang, B.P.; Kim, J.H.; Lee, G.; Kim, H.J.; Lee, J.P.; Yang, C.W.; Kim, Y.S.; Lim, C.S. Pifithrin-α, a p53 inhibitor, stimulates post-ischemia renal fibrosis. Am. J. Physiol. Ren. Physiol. 2015, 308, F578–F587.
57 Lin, S.L.; Kisseleva, T.; Brenner, D.A.; Duffield, J.S. Pericytes and perivascular fibroblasts are the primary source of collagen-producing cells in obstructive fibrosis but not in compensatory hypertrophy following unilateral nephrectomy. Am. J. Pathol. 2008, 173, 1617–1627.
58 Humphreys, B.D.; Valerius, M.T.; McMahon, A.P.; Bonventre, J.V. Wnt1/β-catenin signaling triggers tubular regeneration following acute kidney injury. J. Clin. Investig. 2008, 118, 777–789.
59 Kumar, S.; Gupta, S.; Duffield, J.S. Kidney failure: Developmental pathways in kidney fibrosis. Nature Rev. Nephrol. 2013, 9, 71–73.
60 McCright, B.; Lozier, J.; Gridley, T. Notch signaling in mammalian development and disease. Genes Dev. 2002, 16, 1098–1112.
61 Xiao, L.; Wang, S.; Zhang, X.; Li, H.; Liu, F.; Dong, Z. Notch activation contributes to renal interstitial fibrosis by promoting epithelial-to-mesenchymal transition. Kidney Int. 2015, 87, 547–559.
62 Kim, J.; Ho, J.; Zhang, J.; Chen, L.; Rosner, M.H.; Ronco, C.; Kellum, J.A.; Mehta, R.L.; Martensson, J.; Waikar, S.S.; et al. Notch activation in acute kidney injury. Kidney Int. 2016, 90, 937–947.
63 Onay, T.U.; Elisaf, M.; Goumenos, D.S. The involvement of Notch signaling pathway in acute kidney injury. Ren. Fail. 2016, 38, 1311–1317.
64 Kang, H.M.; Hughes, J.; Park, J.S.; Kugler, P.; Iyer, V.R.; Humphreys, B.D. Distinct fibroblast lineages track kidney nephrogenesis and disease. Cell Stem Cell 2015, 16, 343–355.
65 Kuppe, C.; Ibrahim, M.M.; Kranz, J.; Seeger, F.; Gassler, N.; Kostka, K.; Küppers, F.; Wiggins, J.E.; Gröne, H.J.; Reimer, R.; et al. Decoding myofibroblast origins in human kidney fibrosis. Nat. Med. 2021, 27, 1641–1653.
66 Nakamura, T.; Yamamoto, S.; Sakai, T.; Horikawa, K.; Komori, T.; Sugiyama, T.; Tanaka, T.; Yuzawa, Y.; Shimizu, F.; Taniguchi, M.; et al. Activation of Notch signaling in rat kidney glomeruli in progressive glomerular diseases. J. Am. Soc. Nephrol. 2007, 18, 79–91.
67 Liu, F.; Wang, L.; Ma, L.; Wang, S.; Duffield, J.S. Sonic hedgehog is critical for fibroblast proliferation during kidney fibrosis. Am. J. Pathol. 2004, 164, 393–404.
68 Xie, G.; Liu, F. Role of hedgehog signaling in kidney fibrosis. Fibrogenesis Tissue Repair 2011, 4, 2.
69 Kramann, R.; Schneider, R.K.; Dirocco, D.P.; Machado, F.; Fleig, S.V.; Hafemann, M.; Fogo, A.B.; Duffield, J.S.; Humphreys, B.D. Perivascular Gli1+ progenitors are key contributors to injury-induced organ fibrosis. Cell Stem Cell 2015, 16, 51–66.
70 Schneider, R.K.; Wang, X.; Kramann, R.; Qu, Z.; Humphreys, B.D. Gli2-driven mesenchymal transition of pericytes is a source of activated fibroblasts after acute kidney injury. Cell Stem Cell 2016, 18, 273–286.
71 Xie, G.; Wang, Y.; Liu, F. Inhibition of hedgehog signaling promotes renal tubular cell apoptosis and inhibits tubular cell proliferation. Am. J. Physiol. Ren. Physiol. 2013, 305, F81–F91.
72 He, W.; Dai, C.; Li, Y.; Wolf, G.; Pichler, R.H.; Kumar, R.; Floege, J.; Neilson, E.G.; Fan, J. Wnt/β-catenin signaling promotes renal interstitial fibrosis. I. In vivo and in vitro studies. J. Am. Soc. Nephrol. 2009, 20, 765–781.
73 He, W.; Dai, C.; Li, Y.; Zeng, G.; Wolf, G.; Floege, J.; Schlöndorff, D.; Kumar, R.; Neilson, E.G.; Fan, J. Wnt/β-catenin signaling promotes renal interstitial fibrosis. II. Crosstalk with TGF-β signaling. J. Am. Soc. Nephrol. 2009, 20, 782–790.
74 Katsuno, T.; Isaka, Y. Wnt signaling pathway in kidney diseases. Kidney Int. 2018, 94, 658–665.
75 Lin, S.L.; Chang, D.R.; Chang, A.L.; Fan, L.X.; Sun, Y.; Byrnes, J.R.; Rastaldi, M.P.; Lazzeri, E.; Anders, H.J.; Johnson, T.S.; et al. Macrophage Wnt7b is critical for kidney repair and regeneration following acute kidney injury. Proc. Natl. Acad. Sci. USA 2010, 107, 4194–4199.
76 Wang, Y.; Li, J.; Lin, F.; Zhou, L.; He, X.; Dai, C.; Liu, Y. Canonical Wnt signaling induces nephrogenic mesenchymal cell proliferation and differentiation. J. Am. Soc. Nephrol. 2012, 23, 1875–1885.
77 Kang, H.M.; Huang, S.; Reidy, K.; Han, M.; Breggia, A.; Busse, M.; Berthier, E.; Hastie, A.R.; Wilson, P.C.; Harel, D.; et al. Integrated transcriptomic analysis reveals kidney cell-type specific signatures of chronic kidney disease. J. Am. Soc. Nephrol. 2015, 26, 1666–1682.
78 Surendran, K.; Kim, A.D.; Shankland, S.J. The Wnt/β-catenin signaling pathway in podocytes: A novel target for proteinuria. Kidney Int. 2010, 78, 1178–1186.
79 Wang, Y.; Zhou, L.; He, X.; Li, Y.; Dai, C.; Liu, Y. Canonical Wnt signaling protects against acute kidney injury via promoting tubular cell proliferation. J. Am. Soc. Nephrol. 2013, 24, 1227–1238.
80 Xiao, L.; Wang, S.; Liu, F.; Tang, Y.; Zhang, X.; Wang, J.; Li, H.; Dong, Z. Wnt/β-catenin signaling mediates maladaptive repair and progressive fibrosis after AKI. Kidney Int. 2016, 90, 1135–1146.
81 Zhang, X.; Wang, S.; Xiao, L.; Liu, F.; Zhou, F.; Zhang, L.; Li, H.; Dong, Z. A paradoxical role of Wnt/β-catenin signaling in kidney repair after AKI. J. Am. Soc. Nephrol. 2018, 29, 51–65.
82 Haase, V.H. Hypoxia-inducible factors in chronic kidney disease. Kidney Int. Suppl. 2013, 3, 401–404.
83 Poelstra, K.; Bakker, J.; Verhaar, M.C.; Reutelingsperger, C.P.M.; van Zonneveld, A.J.; Claessen, N. The protective role of hypoxia-inducible factor-1α in acute kidney injury is independent of tubular epithelial cells. Kidney Int. 2013, 83, 621–630.
84 Poelstra, K.; Bakker, J.; Verhaar, M.C.; Reutelingsperger, C.P.M.; van Zonneveld, A.J.; Claessen, N. The protective role of hypoxia-inducible factor-1α in acute kidney injury is independent of tubular epithelial cells. Kidney Int. 2013, 83, 621–630.
85 Bernhardt, W.M.; Campean, V.; Kany, S.; Juergensen, J.S.; Weidemann, A.; Warnecke, C.; Wiesener, M.S.; Eckardt, K.U. Preischemic stabilization of hypoxia-inducible factor prevents chronic progressive renal failure after acute ischemic injury. Kidney Int. 2009, 75, 642–653.
86 Kalluri, R.; Weinberg, J.M. Targeting fibrosis in chronic kidney disease. J. Am. Soc. Nephrol. 2010, 21, 1213–1220.
87 Kimura, H.; Weihbrecht, M.; Wagner, H.J.; Warnecke, C.; Bernhardt, W.M.; Wiesener, M.S.; Eckardt, K.U. Cell-type-specific roles of hypoxia-inducible factor-2α in the kidney. Kidney Int. 2008, 73, 409–415.
88 Liu, X.; Zhou, F.; Zhang, X.; Wang, S.; Xiao, L.; Zhang, L.; Li, H.; Dong, Z. Hypoxia-inducible factor-1α is dispensable for kidney repair after ischemic acute kidney injury. Am. J. Pathol. 2016, 186, 2693–2704.
89 Rosenberger, C.; Mandriani, B.; Bernhardt, W.M.; Wiesener, M.S.; Brüne, B.; Eckardt, K.U. Hypoxia inducible factor-1α is a regulator of migration and proliferation of renal tubular cells in vitro. Kidney Int. 2004, 66, 2400–2411.
90 Tanaka, T.; Nangaku, M. Hypoxia and kidney fibrosis: The role of hypoxia-inducible factor. Kidney Int. Suppl. 2013, 3, 105–109.
91 Liu, Y.; Ramsubir, S.; Lee, I.S.; Choi, M.E. Hypoxia-inducible factor 1α mediates transforming growth factor-β1-induced expression of extracellular matrix proteins in tubular epithelial cells: Involvement of Smad3. J. Biol. Chem. 2003, 278, 43679–43685.
92 Bonventre, J.V.; Zuk, A. Bax and Bak in kidney injury and repair. Kidney Int. 2004, 66, 2214–2217.
93 Jiang, M.; Wei, Q.; Dong, G.; Ryu, D.R.; Yin, Y.; Potenza, M.A.; Zhang, W.; Lieberthal, W.; Feng, Y.; Dong, Z. Bax and Bak Mediate Cisplatin-Induced Mitochondrial Outer Membrane Permeabilization and Apoptosis. J. Biol. Chem. 2014, 289, 2071–2084.
94 Youle, R.J.; van der Bliek, A.M. Mitochondrial fission, fusion, and stress. Science 2012, 337, 1062–1065.
95 Suen, D.F.; Norris, K.L.; Youle, R.J. Mitochondrial fission and fusion in health and disease. Trends Cell Biol. 2008, 18, 362–371.
96 Dong, Z.; Wei, Q.; Xu, C.; Wei, Y.; Liu, F.; Bhatia, D.; Cao, G.; Yang, J.; Kumar, R.; Du, Y.; et al. The pathogenic role of mitochondrial fission in kidney disease. J. Am. Soc. Nephrol. 2017, 28, 2080–2097.
97 Choi, J.A.; Tao, M.; Kim, J.; Kim, J.H.; Ham, S.; Chung, H.K.; Lee, J.E.; Joo, K.W.; Oh, K.H.; Kim, Y.S.; et al. Drp1 inhibition protects against cisplatin-induced acute kidney injury through the modulation of mitochondrial fission and apoptosis. Nephrol. Dial. Transplant. 2018, 33, 1308–1318.
98 Wei, Q.; Dong, G.; Zhang, J.; Shao, X.; Yang, L.; Miao, L.; Liu, X.; Zhang, L.; Wang, Y.; Liu, F.; et al. Mdivi-1 protects against cisplatin-induced nephrotoxicity by preventing mitochondrial fission. J. Am. Soc. Nephrol. 2015, 26, 1904–1915.
99 Linkermann, A.; Stockwell, B.R.; Krautwald, S.; Anders, H.J. Regulated cell death and inflammation: An auto-amplifying loop causes organ damage. Nat. Rev. Immunol. 2014, 14, 759–767.
100 Baines, C.P. The mitochondrial permeability transition pore and ischemia-reperfusion injury. Basic Res. Cardiol. 2009, 104, 181–195.
101 Basile, D.P.; Anderson, M.D.; Davis, J.A. Role of the mitochondrial permeability transition pore in tubular cell death in ischemic acute kidney injury. Am. J. Physiol. Ren. Physiol. 2016, 310, F1225–F1234.
102 Dhall, D.; Mittal, A.; Kumar, V.; Lall, S.B.; Pathak, R.; Bhatnagar, A.; Sharma, K.; Sharma, R. Kidney mitochondrial dysfunction in chronic kidney disease. J. Am. Soc. Nephrol. 2020, 31, 1621–1636.
103 Szeto, H.H. Mitochondria-targeted peptide antioxidants for ischemia-reperfusion injury. Antioxid. Redox Signal. 2008, 10, 1611–1631.
104 Susztak, K.; Raff, A.C.; Schiffer, M.; Müller-Decker, K.; Singh, M.K.; Satirapoj, B.; Nelson, R.G.; Mäser, P.; Derfler, K.; Huber, T.M.; et al. Hyperglycemia induces a switch to fatty acid β-oxidation and lipid deposition in renal tubular cells. Am. J. Pathol. 2007, 171, 406–418.
105 Bhargava, P.; Schnellmann, R.G. Mitochondrial energetics, dynamics, and signaling in acute kidney injury. Kidney Int. 2017, 91, 1042–1056.
106 Las, G.I.; Serban, C.; Protzer, P.; Haller, H.; Dumler, I.; Mertens, P.R. High glucose-induced mitochondrial fission and dysfunction in kidney tubular cells is mediated via dynamin-related protein-1. J. Biol. Chem. 2018, 293, 5738–5753.
107 Sharma, K.; Karl, B.; Mathew, A.V.; Gounder, S.S.; Pillai, R.; Gubbi, S.; Gobe, G.; Campbell, P.D.; Ramm, G.; Grey, S.T.; et al. Metabolomics of human diabetic kidney disease. J. Clin. Investig. 2013, 123, 5354–5363.
108 Ramesh, G.; Schnellmann, R.G. Activating peroxisome proliferator-activated receptor γ enhances mitochondrial biogenesis and decreases cisplatin-induced proximal tubule apoptosis. J. Pharmacol. Exp. Ther. 2009, 328, 940–948.
109 Ramesh, G.; Schnellmann, R.G. PPARγ agonists as therapeutics for acute kidney injury. Kidney Int. 2010, 77, 394–399.
110 Ramesh, G.; Wei, Q.; Reeve, J.L.; Schnellmann, R.G. Activating peroxisome proliferator-activated receptor γ stimulates mitochondrial biogenesis in cisplatin-induced acute kidney injury. Am. J. Physiol. Ren. Physiol. 2013, 304, F14–F22.
111 Wei, Q.; Ramesh, G.; Schnellmann, R.G., Activating mitochondrial biogenesis protects against cisplatin-induced acute kidney injury. J. Am. Soc. Nephrol. 2013, 24, 568–579.
112 Parikh, S.M.;хваতিয়ে, R.; Inzucchi, S.E.; Young, L.H.; Orlando, R.A. PGC-1α is required for mitochondrial biogenesis in the murine kidney and is dispensable for renal function. Am. J. Physiol. Ren. Physiol. 2009, 296, F1378–F1387.
113 Oberg, B.P.; McMenamin, E.; Lucas, F.L.; McMonagle, E.; Morrow, J.; Ikizler, T.A.; Himmelfarb, J. Increased prevalence of oxidant stress and inflammation in patients with moderate to severe chronic kidney disease. Kidney Int. 2004, 65, 1009–1016.
114 Ramos, A.M.; Luft, V.C.; Watanabe, P.M.; Camargo, R.L.; Dalboni, M.A.; Seguro, A.C. Oxidative stress and body composition in chronic kidney disease patients. J. Bras. Nefrol. 2012, 34, 225–232.
115 Fedorova, O.; Goncharov, N.; Mzhavia, N.; Staruschenko, A. Autophagy and mitochondrial biogenesis in experimental chronic kidney disease. PLoS ONE 2012, 7, e48255.
116 Yao, Y.; Wang, L.; Chun, J.; Chen, Y.; Gonzalez, F.J.; Megyesi, J.K.; Safirstein, R.L. Nrf2 protects against ischemic kidney injury via induction of antioxidant genes. J. Am. Soc. Nephrol. 2012, 23, 1932–1943.
117 Himmelfarb, J.; McMonagle, E.; McMenamin, E.; Freedman, S.; Leucker, T.; Ikizler, T.A.; Hakim, R.M.; Miller, B.G.; Paganini, E.P.; Charytan, C.; et al. Plasma protein oxidation in patients with acute renal failure. Kidney Int. 2002, 61, 413–423.
118 Zhang, Y.; Chen, J.; Zhao, Y.; Liu, X.; Wang, Y.; Li, Y.; Liu, F.; Dong, Z. Obesity exacerbates trauma-induced acute kidney injury by increasing oxidative stress and inflammation. Am. J. Physiol. Ren. Physiol. 2018, 315, F137–F147.
119 McCullough, P.A.; Choi, J.P.; Haller, S.T.; Wolyn, R.; Bakris, G.L.; Forman, M.B. Contrast-induced acute kidney injury after coronary angiography: Risk factors and outcomes. J. Am. Coll. Cardiol. 2000, 36, 2272–2278.
120 Toprak, O.; Cirit, M.; Tanrisevdi, H.; Uygur, F.; Canbakan, M.; Ozbayoglu, O.; Onat, A.; Vanholder, R.; Akpolat, T. Predictors of contrast-induced nephropathy: A prospective multi-center study. Nephrol. Dial. Transplant. 2007, 22, 477–484.
121 Heyman, S.N.; Rosenberger, C.; Rosen, S.R. Contrast nephropathy. Clin. J. Am. Soc. Nephrol. 2016, 11, 326–334.
122 Kaushal, G.P.; Kaushal, V. Autophagy in acute kidney injury and chronic kidney disease. Adv. Exp. Med. Biol. 2019, 1165, 455–480.
123 Jiang, M.; Liu, Y.; Xie, Y.; Shao, X.; Zhang, J.; Zhang, L.; Wang, Y.; Liu, F.; Dong, Z. Ruboxistaurin protects against ischemic acute kidney injury by inducing autophagy via activation of the AMPK pathway. Free Radic. Biol. Med. 2016, 97, 368–381.
124 Jiang, M.; Wei, Q.; Zhang, J.; Dong, G.; Shao, X.; Zhang, L.; Wang, Y.; Liu, F.; Dong, Z. Endoplasmic reticulum stress-induced autophagy protects against tunicamycin-induced acute kidney injury. Cell Death Dis. 2016, 7, e2226.
125 Kimura, T.; Takabatake, Y.; Takahashi, A.; Yamamoto, T.; Matsui, T.; Kobori, H.; Okada, H.; Yamamoto, K.; Namba, T.; Katagiri, D.; et al. Autophagy protects the proximal tubule from degeneration and acute kidney injury. J. Am. Soc. Nephrol. 2011, 22, 1790–1800.
126 Ko, A.; Nishikawa, M.; Kiriyama, T.; Miyata, T.; Kawashima, A.; Tanaka, S.; Tamura, K.; Kimura, K.; Nakada, C.; Arii, J.; et al. Rapamycin alleviates renal tubular cell injury in vitro and in vivo via autophagy induction. Nephrol. Dial. Transplant. 2011, 26, 3432–3440.
127 Ravichandran, K.; Baisantry, A.; Wang, Z.V.; Ballou, L.R.; Choudhury, G.G.; McMullen, J.R.; Hill, J.A.; Jaimes, E.A.; Johnson, A.C.; Chung, M.K.; et al. Beclin 1 haploinsufficiency increases infarct size and postischemic cardiac dysfunction in mice. J. Clin. Investig. 2009, 119, 2882–2892.
128 Takahashi, A.; Takabatake, Y.; Kimura, T.; Matsui, T.; Namba, T.; Saka, Y.; Kawabata, K.; Kaimori, J.Y.; Kitamura, M.; Matsusaka, T.; et al. Autophagy in the proximal tubule is sexually dimorphic and displays differential time course after acute kidney injury. Autophagy 2012, 8, 1503–1513.
129 Baisantry, A.; Choudhury, G.G.; Ravichandran, K.; Wang, J.; Williams, J.; Dong, Z.; Nickerson, M.L.; Humphreys, B.D.; Livingston, M.J.; Prasad, P.V. Renal proximal tubule-specific deletion of autophagy protein Atg5 promotes kidney injury but suppresses fibrosis. J. Biol. Chem. 2016, 291, 7821–7835.
130 Livingston, M.J.; Baisantry, A.; Nickerson, M.L.; Williams, J.; Choudhury, G.G.; Jaimes, E.A.; Johnson, A.C.; Wang, Z.V.; Prasad, P.V. Renal proximal tubule autophagy mediates protection against chronic kidney disease. Autophagy 2016, 12, 143–156.
131 Li, J.; Sharma, V.M.; Qu, Y.; Singh, K.; Watkins, T.; Szalai, A.J.; Zhou, G.; Kumar, R.; Bhatia, D.; Dong, Z.; et al. Specific deletion of autophagy in proximal tubules protects against chronic kidney disease. Cell Death Dis. 2020, 11, 324.
132 Lenoir, O.; Tharaux, P.L. Nutrient sensors in diabetic nephropathy: The role of mTORC1 and AMPK. Kidney Int. 2015, 88, 462–475.
133 Srivastava, S.P.; Roy Choudhury, G.; Bhardwaj, R.; Sharma, R.K.; Tripathi, M.; Singh, R.K.; Jha, V.; Prasad, P.V. Defective autophagy in podocytes accelerates diabetic nephropathy. Kidney Int. 2015, 87, 945–957.
134 Tang, C.; Liu, Y.; Hu, J.; Bi, X.; Zhang, L.; Li, Q.; Liu, H.; Guo, L.; Yan, M.; Wang, F.; et al. SIRT1/PGC-1α pathway activation by SRT1720 attenuates diabetic nephropathy via autophagy enhancement and oxidative stress and inflammation suppression. Oxid. Med. Cell. Longev. 2016, 2016, 9583458.
135 Galluzzi, L.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cecconi, F.; Choi, A.M.K.; Chu, C.T.; Codogno, P.; Colombo, M.I.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222.
136 Lee, J.H.; Kim, J.; Lee, M.J.; Park, S.K.; Kim, H.J.; Song, J.; Lee, M.K.; Lee, H.E.; Chung, C.H. Autophagy deficiency may potentiate endoplasmic reticulum stress and cell death in the kidneys of type 1 diabetic rats. PLoS ONE 2014, 9, e98796.
137 Hoshino, A.; Kikuchi, R.; Watanabe, R.; Gomi, F.; Tanaka, M.; Aramaki, Y.; Ishihara, K.; Gotoh, N.; Shibata, T.; Kawakami, H.; et al. Loss of SQSTM1/p62 in podocytes exacerbates diabetic nephropathy. Sci. Rep. 2016, 6, 31627.
138 Yamahara, K.; Yasuda, M.; Namba, T.; Tanaka, Y.; Saka, Y.; Kobayashi, T.; Hamano, T.; Nakatani, T.; Okada, A.; Takeda, Y.; et al. Decreased renal tubular cell autophagy in human diabetic nephropathy. Nephrol. Dial. Transplant. 2013, 28, 349–358.
139 Kim, S.J.; Lee, J.H.; Lee, J.P.; Oh, Y.K.; Kim, Y.S.; Lim, C.S. Mammalian target of rapamycin complex 1 activation is associated with autophagy reduction in podocytes of focal segmental glomerulosclerosis patients. Nephrology 2015, 20, 249–257.
140 Li, H.; Liu, Y.; Zhang, Y.; Wang, J.; Zhao, Y.; Li, Y.; Liu, F.; Dong, Z. Defective autophagy in aged kidneys impairs cellular homeostasis and increases tissue injury. Aging Cell 2017, 16, 62–75.
141 Es, S.V.; Guermonprez, P.; Vallat, C.; Desoutter, J.F.; Védie, A.L.; Aucouturier, P.; Ronco, P.; Verollet, G. Autophagy is induced in human glomerulonephritis. PLoS ONE 2011, 6, e19086.
142 Humphreys, B.D. Acute kidney injury—Recovery after injury. Semin. Nephrol. 2019, 39, 261–265.
143 Sharifian, M.; Elhmidi, Y.; Mirza, M.; Zhang, J.; Chan, L.; Yang, W.; Lee, H.T.; Yang, L.; Park, P.J.; Molitoris, B.A.; et al. A transcriptomic atlas of mouse kidney injury and repair. J. Am. Soc. Nephrol. 2022, 33, 201–218.
144 Venkatachalam, M.A.; Patel, Y.V. AKI-CKD transition: Is maladaptive repair the culprit? Am. J. Physiol. Ren. Physiol. 2020, 319, F617–F627.
145 Tonelli, M.; Sacks, F.M.; Pfeffer, M.A.; Gao, Z.; Curhan, G.C. Biomarkers of inflammation and progression of chronic kidney disease. Kidney Int. 2005, 68, 237–245.
146 Amdur, R.L.; Mehta, S.; Navaneethan, S.D.; Nally, J.V.; Fischer, M.J.; Mohanty, K.; Fink, N.E.; Porter, A.C.; O’Hare, A.M.; Lash, J.P.; et al. Association of plasma tumor necrosis factor-α with rapid kidney function decline in chronic kidney disease: The chronic renal insufficiency cohort (CRIC) study. Nephrol. Dial. Transplant. 2016, 31, 1841–1849.
147 Lee, J.S.; Lee, C.H.; Lee, J.H.; Kim, S.H.; Lee, H.W.; Lee, J.H.; Kim, K.L.; Park, K.S.; Kim, Y.S.; Lee, J.P.; et al. Preoperative chronic kidney disease as a predictor of postoperative infection, acute kidney injury, and in-hospital death after major surgery. Medicine 2016, 95, e4428.
148 Bostom, A.G. Homocysteine and chronic kidney disease: Chicken or egg? Nephrol. Dial. Transplant. 2003, 18, 2426–2429.
149 Kaysen, G.A. Role of inflammation and nutritional status in chronic kidney disease. Adv. Chronic Kidney Dis. 2005, 12, 135–145.
150 Stenvinkel, P. Chronic kidney disease inflammation—Full circle? Nephrol. Dial. Transplant. 2017, 32, 201–203.
151 Stenvinkel, P.; Heimburger, O.; Lönnqvist, F.; Bergman, P.; Tranaeus, A.; Füllgrabe, H.; Axelsson, J.; Harris, R.A.; Endert, E.; Hulting, J.; et al. Circulating levels of interleukin-10 are associated with decreased fat mass in chronic kidney disease. J. Am. Soc. Nephrol. 2003, 14, 193–199.
152 Carrero, J.J.; Stenvinkel, P. Inflammation in end-stage renal disease—What have we learned and where do we go from here? Semin. Dial. 2010, 23, 498–509.
153 Girndt, M.; Sester, U.; Sester, M.; Köhler, H. Serum levels of tumor necrosis factor-α and interleukin-6 in chronic renal diseases: Potential significance for activation of coagulation and stimulation of lipogenesis. Kidney Int. 1997, 51, 1785–1791.
154 Honda, T.; Nangaku, M. Inflammation in acute kidney injury. Kidney Dis. 2016, 2, 108–112.
155 Liu, Y.; Cao, X. TWEAK/Fn14 pathway in kidney diseases. Kidney Dis. 2016, 2, 119–127.
156 Vlasschaert, J.L.; D’Haese, P.C. The role of interleukin-6 in chronic kidney disease. Kidney Int. 2018, 94, 866–878.
157 Hu, M.C.; Shi, M.; Zhang, J.; Qin, L.; Dai, J.; Wu, J.; Li, C.; Chen, C.; Oriste, P.; Huang, J.; et al. Klotho and acute kidney injury. Kidney Int. 2015, 88, 720–727.
158 Schrier, R.W.; Wang, W.; Poole, B.; Mitra, A.; Chan, L.; Sharma, K. Acute renal failure: Definitions, diagnosis, pathogenesis, and therapy. J. Am. Soc. Nephrol. 2004, 15, 1532–1543.
159 Liu, Y.; Chen, Y.; Han, Z.; Liu, F.; Cao, X. TWEAK-Fn14 signaling promotes pericyte-mediated inflammation and vasoconstriction in acute kidney injury. J. Am. Soc. Nephrol. 2017, 28, 78–91.
160 Choudhury, G.G.; Nickerson, M.L.; Williams, J.; Baisantry, A.; Neilson, E.G.; Pazour, G.J.; Prasad, P.V. Polycystin-1 regulates renal tubular epithelial cell proliferation and apoptosis via STAT3 signaling. Hum. Mol. Genet. 2013, 22, 1312–1325.
161 Choudhury, G.G.; Williams, J.; Nickerson, M.L.; Baisantry, A.; Prasad, P.V. Activation of signal transducer and activator of transcription 3 in polycystic kidney disease. J. Am. Soc. Nephrol. 2010, 21, 1613–1625.
162 Ospina-Tascon, G.A.; Bautista-Rincón, D.F.; Umana-Mendoza, M.; Vargas-Pabon, M.; Rodriguez-Malagon, N.; Bermúdez, C.; Pérez, A.; Sánchez-Ortiz, A.I.; García, A.F.; Vincent, J.L.; et al. Sepsis-associated acute kidney injury: Current knowledge and future directions. Biomedica 2019, 39, 679–705.
163 Bruchfeld, A.; Stenvinkel, P.; Qureshi, A.R.; Barreto, D.V.; Linde, T.; Lindholm, B.; Lundin, G.; Heimbürger, O. Serum levels of high mobility group box-1 protein are elevated in patients with chronic kidney disease. Nephrol. Dial. Transplant. 2008, 23, 2193–2197.
164 Andersson, U.; Tracey, K.J. HMGB1 is a therapeutic target for sterile inflammation and infection. Annu. Rev. Immunol. 2011, 29, 139–162.
165 Andersson, U.; Wang, H.; Palmblad, K.; Ave, P.; Bloom, O.; Erlandsson-Harris, H.; Janson, A.; Kokkola, R.; Li, J.; Moro, V.; et al. High mobility group 1 protein (HMGB1) stimulates proinflammatory cytokine synthesis in macrophages. J. Exp. Med. 2000, 192, 565–570.
166 Awad, A.S.; Huang, Q.; Sequeira, V.; Fallows, J.; Brennand, C.; Hamilton, J.A.; Yaqoob, M.M. High-mobility group box 1 protein is a novel mediator of inflammation in ischaemic acute kidney injury. Nephrol. Dial. Transplant. 2011, 26, 1986–1996.
167 Kim, S.; Park, S.Y.; Lee, J.H.; Lee, J.P.; Choi, H.J.; Kim, Y.S.; Lim, C.S.; Kim, Y.L. Recombinant human high-mobility group box 1 protein induces renal tubular cell apoptosis in vitro and acute kidney injury in vivo. Am. J. Physiol. Ren. Physiol. 2015, 309, F809–F818.
168 Star, R.A.; Hogan, S.L.; Illei, G.G.; Racusen, L.C.; Imam, S.Z.; Kelly, C.J.; Humphreys, B.D.; Bonventre, J.V.; Gill, N.; Nzerue, C.M.; et al. Kidney disease, high-mobility group box-1 protein, and systemic inflammatory diseases. J. Am. Soc. Nephrol. 2012, 23, 1070–1081.
169 Ascensão, R.; Gonçalves, J.; Mendes, V.; Oliveira, J.; Sereno, J.; Teixeira, M.; Magalhães, J.; Neves, J.; Carvalho, A.M.; Oliveira, C.; et al. Association between interleukin-8 and renal function, inflammation and oxidative stress in chronic kidney disease patients. Clin. Nephrol. 2013, 79, 462–468.
170 Barreto, D.V.; Barreto, F.C.; Carvalho, A.B.; Vieira, M.L.; Liabeuf, S.; Meert, N.; Massy, Z.A.; Choukroun, G.; Drueke, T.B.; Cendoroglo, M.; et al. Association of interleukin-18 and interferon-γ with mortality in hemodialysis patients. J. Am. Soc. Nephrol. 2010, 21, 127–134.
171 Swardfager, W.; Au, B.; MacNeil, M.L.; Herrmann, N.; Andreou, C.; Steinberg, G.R.; Lanctôt, K.L. Systemic interferon signaling in chronic kidney disease: Associations with neurocognitive function. J. Neuroinflammation 2019, 16, 23.
172 Wynn, T.A.; Vannella, K.M. Macrophage biology in development, homeostasis and disease. Nature 2016, 529, 172–179.
173 Wynn, T.A.; Chawla, A.; Pollard, J.W. Macrophage biology in development, homeostasis and disease. Nature 2013, 496, 445–455.
174 Li, L.; Huang, L.; Sung, S.J.; Lobo, P.I.; Gee, M.S.; Heeger, P.S.; Lee, H.T. Macrophages recover kidney from acute injury. Kidney Int. 2008, 73, 394–402.
175 Anders, H.J.; Vielhauer, V. Renal fibrosis and the inflammatory infiltrate. Kidney Int. 2004, 66, 419–431.
176 Hesse, A.; Kretzler, M. Macrophages in the kidney: Lessons from murine models of renal disease. Kidney Int. 2016, 90, 948–961.
177 Ricardo, S.D.; van Goor, H.; Eddy, A.A. Macrophage diversity in renal injury and repair. J. Am. Soc. Nephrol. 2008, 19, 1257–1268.
178 London, G.M.; Marchais, S.J.; Guerin, A.P.; Metivier, F.; Pannier, B.; Boudouin, B.; Levy, B.I. Arteriosclerosis, vascular calcifications and cardiovascular disease in uremic patients. Curr. Opin. Nephrol. Hypertens. 2000, 9, 703–709.
179 Stehouwer, C.D.A.; Gall, M.A.; Twisk, J.W.R.; Knudsen, E.; Emeis, J.J.; Parving, H.H. Increased urinary albumin excretion, endothelial dysfunction, and chronic low-grade inflammation in type 2 diabetes: Progressive, interrelated, and independently associated with risk of death. Diabetes 2002, 51, 1157–1165.
180 Yilmaz, M.I.; Carrigan, T.P.; Jr; de Ca