Introduction
Arthrogryposis–renal dysfunction–cholestasis (ARC) syndrome (MIM 208085) is a rare, life-threatening autosomal recessive disorder that presents significant diagnostic challenges. First recognized in 1973 in offspring from a consanguineous marriage [1], ARC syndrome is characterized by a constellation of clinical features affecting multiple systems, including musculoskeletal, renal, hepatic, and central nervous systems. Accurate and timely Arc Diagnosis is crucial for appropriate management and genetic counseling, yet the rarity and variable presentation of the syndrome often lead to delays or misdiagnosis. The hallmark features of ARC syndrome include arthrogryposis, renal tubular acidosis, and neonatal cholestatic jaundice (Figure 1) [2]. These core features are frequently accompanied by other manifestations such as ichthyosis (in approximately 50% of cases), platelet anomalies (around 25%), agenesis of the corpus callosum (greater than 20%), congenital cardiovascular anomalies (about 10%), deafness, recurrent infections, and bleeding tendencies due to coagulation dysfunction (Table 1). Laboratory investigations and organ biopsies of the liver or kidney play a vital role in confirming the arc diagnosis. However, subtle or atypical initial symptoms can easily lead to overlooking this severe condition, resulting in delayed treatment and contributing to the poor prognosis, with most patients not surviving beyond their first year of life [3, 4].
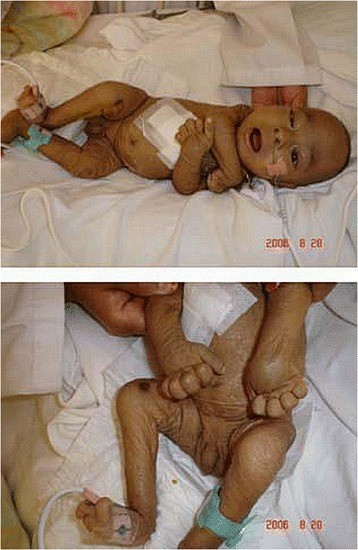 An infant with ARC syndrome showing arthrogryposis and ichthyotic skin.
An infant with ARC syndrome showing arthrogryposis and ichthyotic skin.
Clinical Presentation of ARC Syndrome: Key Diagnostic Indicators
The clinical presentation of ARC syndrome is multifaceted, requiring a comprehensive approach to arc diagnosis. Recognizing the key clinical indicators across different organ systems is essential for early suspicion and confirmation.
Arthrogryposis as a Diagnostic Feature
Arthrogryposis, a congenital joint contracture in two or more body areas, is a primary diagnostic symptom of ARC syndrome. It manifests as a spectrum of musculoskeletal abnormalities, including muscle atrophy, radial deviation of the wrist, dislocations of the hip joints bilaterally, flexion contractures of the knee joints, and calcaneovalgus [2]. It is important to note that musculoskeletal abnormalities might not be immediately apparent in the first few weeks of life, or they may present atypically in certain VPS33B mutations, such as 971delA/K324fs [16, 17]. While the underlying pathogenesis involves the degeneration of anterior motor neurons, the severity of arthrogryposis can be influenced by placental insufficiency and oligohydramnios during pregnancy, leading to fetal growth restriction. The presence of osteopenia and pathological fractures in ARC syndrome is linked to impaired renal tubular reabsorption and secondary hyperparathyroidism. Therefore, in newborns presenting with arthrogryposis alongside osteopenia or fractures, ARC syndrome should be considered in the differential arc diagnosis, especially when accompanied by other clinical features such as renal tubular dysfunction and cholestasis.
Renal Tubular Dysfunction: A Critical Component of ARC Diagnosis
Renal tubular dysfunction, often manifesting as Fanconi syndrome, is another cornerstone in the arc diagnosis of ARC syndrome. Symptoms include renal tubular acidosis, nephrogenic diabetes insipidus, glucosuria, aminoaciduria, and phosphaturia [19, 20]. Renal tubular acidosis can be significantly exacerbated during intercurrent illnesses, indicative of renal tubular calcification and degeneration. Renal ultrasonography may reveal nephrocalcinosis or small dysplastic kidneys. Renal biopsy findings often include inflammatory reactions in the renal interstitium, focal glomerulosclerosis, and tubular distortion and degeneration [3, 16]. These renal manifestations are crucial for differentiating ARC syndrome from other causes of neonatal cholestasis and arthrogryposis.
Neonatal Cholestatic Jaundice: Differentiating ARC Syndrome
Neonatal cholestatic jaundice, frequently accompanied by hepatomegaly, is the third cardinal feature of ARC syndrome and a vital clue for arc diagnosis. The cholestasis observed in ARC syndrome has distinct characteristics compared to other causes of neonatal jaundice. Patients with ARC syndrome typically present with jaundice and liver cell dysfunction but without biliary obstruction. A key differentiating factor is consistently low γ-glutamyl transpeptidase (γGT) levels, along with normal or mildly elevated aspartate aminotransferase (AST) and alanine aminotransferase (ALT) levels [17]. Low GGT cholestasis is a significant indicator for arc diagnosis, particularly when associated with other ARC syndrome features. In cases of conjugated hyperbilirubinemia with low-GGT, especially when accompanied by ichthyosis, deafness, platelet dysfunction, and central nervous system malformations, VPS33B-related disease should be strongly suspected [17]. Liver biopsies in these patients often reveal a paucity of bile ducts, giant cell transformation, bile plugs or lipofuscin deposition, and portal fibrosis, which helps to specifically exclude biliary atresia [3, 21, 22], further refining the arc diagnosis.
Other Associated Features: Expanding the Diagnostic Spectrum
Beyond the classical triad, several additional clinical symptoms can contribute to a comprehensive arc diagnosis. These include ichthyosis, platelet abnormalities, recurrent infections, and cardiovascular anomalies [3, 9].
-
Ichthyosis: Affecting a majority of patients, ichthyosis in ARC syndrome arises from defects in SNARE proteins, which are crucial for epidermal cohesion and the waterproofing function of lamellar granules [23, 24]. Skin biopsies may show mild hyperkeratosis without parakeratosis.
-
Platelet Abnormalities: While routine platelet counts may be normal, patients with ARC syndrome can experience self-limiting intra-abdominal hemorrhages due to abnormal platelet function [9]. Despite an increased number of β-granules, similar to Grey platelet syndrome, platelets exhibit abnormal biosynthesis and function of α-granules, essential for platelet aggregation and thrombogenesis [26, 27].
-
Recurrent Infections: Patients often suffer from recurrent secondary infections, hyperpyrexia, and chronic diarrhea, despite having normal immunological profiles. This increased susceptibility to infections is linked to a defect in phagosome-lysosome fusion caused by Vps16B/Vps33B dysfunction [30].
-
Cardiovascular Anomalies: Congenital cardiovascular anomalies can also be present in ARC syndrome, sometimes leading to bacterial endocarditis secondary to recurrent infections [3].
Genetic Basis of ARC Syndrome: Confirming the Arc Diagnosis
The genetic underpinnings of ARC syndrome are crucial for confirming the clinical arc diagnosis and providing accurate genetic counseling. The disorder is linked to germline mutations in two genes: vacuolar protein sorting 33 homolog B (VPS33B; MIM 608552) and VPS33B-interacting protein, apical–basolateral polarity regulator (VIPAR; MIM 613401) [3, 5, 6].
VPS33B mutations are found in approximately 75% of clinically diagnosed ARC syndrome cases [9]. This gene encodes a protein essential for intracellular vesicular trafficking pathways [7]. Mutations in VPS33B disrupt the interaction between the expressed mutant protein and SNARE proteins, leading to abnormal localization of plasma proteins in polarized cells, contributing to the pathophysiology of ARC syndrome.
VIPAR (C14ORF133) mutations account for the remaining cases of ARC syndrome. VIPAR interacts with VPS33B, and the VPS33B-VIPAR complex plays a role in RAB11A-dependent apical recycling and the regulation of adherent proteins like E-cadherin, essential for cellular polarity [6, 10]. Dysfunction of this complex disrupts organelle biogenesis, affecting tissues like bile ducts and renal tubules, resulting in cholestasis and renal dysfunction, characteristic features of ARC syndrome.
To facilitate arc diagnosis and research, the Leiden Open-Source Variation Database (LOVD) for ARC (https://grenada.lumc.nl/LOVD2/ARC) was established in 2011 [31]. As of March 2014, this database compiled 299 unique VPS33B variants and 34 VIPAR variants, classified by their projected pathogenicity. Currently, the database lists 49 pathogenic VPS33B and 14 pathogenic VIPAR mutations worldwide. The most frequent pathogenic VPS33B variants include c.403 + 2 T > A, c.1312C > T, and c.1519C > T (Table 2), while recurrent pathogenic VIPAR variants are c.658C > T and c.808C > T (Table 3). This genetic information is invaluable for clinicians in confirming arc diagnosis, understanding prognosis, and providing genetic counseling.
Diagnostic Workup for ARC Syndrome
The arc diagnosis of ARC syndrome requires a multi-pronged approach, integrating clinical evaluation, laboratory investigations, and genetic testing.
Clinical Evaluation
A thorough clinical evaluation is the first step in arc diagnosis. This includes:
- Detailed Family History: Consanguinity increases the risk of autosomal recessive disorders like ARC syndrome.
- Assessment of Cardinal Features: Evaluating for arthrogryposis, neonatal cholestatic jaundice, and renal tubular dysfunction.
- Identification of Associated Features: Checking for ichthyosis, platelet abnormalities, deafness, and other symptoms.
Laboratory Investigations
Laboratory tests are crucial for supporting the clinical suspicion and refining the arc diagnosis:
- Liver Function Tests: Assessing bilirubin levels, AST, ALT, and γGT. Low γGT in the context of cholestasis is a significant diagnostic clue.
- Renal Function Tests: Evaluating electrolytes, blood pH, bicarbonate levels to detect renal tubular acidosis, and urine analysis for glucosuria, aminoaciduria, and phosphaturia.
- Platelet Function Tests: While routine platelet counts may be normal, specialized platelet function tests may reveal abnormalities.
- Renal and Liver Ultrasound: Imaging to detect nephrocalcinosis, dysplastic kidneys, or hepatomegaly.
- Liver and Renal Biopsies: Histopathological examination can reveal characteristic features such as paucity of bile ducts, giant cell transformation in the liver, and tubular degeneration and nephrocalcinosis in the kidney. However, biopsies carry a risk of bleeding in these patients.
Genetic Testing: The Definitive Arc Diagnosis Tool
Genetic testing for VPS33B and VIPAR mutations is the gold standard for confirming arc diagnosis. Sequencing analysis can identify pathogenic mutations in these genes. While mutational analysis is highly specific, it can be time-consuming and may occasionally yield false negatives. In such cases, alternative diagnostic techniques like VPS33B protein expression analysis in skin fibroblasts and platelet morphology assessment in peripheral blood smears can be valuable supplementary tools [9, 36].
Differential Diagnosis
The arc diagnosis requires careful differentiation from other conditions presenting with similar features, especially neonatal cholestasis, arthrogryposis, and renal dysfunction. Conditions to consider include:
- Biliary Atresia: Distinguished by the presence of biliary obstruction, typically with elevated γGT levels, which are low in ARC syndrome. Liver biopsy findings are also distinct.
- Progressive Familial Intrahepatic Cholestasis (PFIC): Shares some clinical and laboratory features with ARC syndrome. Genetic testing is essential for differentiation.
- Cerebro-oculo-facio-skeletal (COFS) syndrome: Features arthrogryposis and neurological abnormalities, but typically lacks the renal and cholestatic features of ARC syndrome.
- Other causes of neonatal cholestasis: Including infections, metabolic disorders, and endocrine disorders. A comprehensive clinical and laboratory evaluation is necessary to distinguish these from ARC syndrome.
Treatment and Prognosis: Implications for Arc Diagnosis
Currently, there is no specific cure for ARC syndrome. Treatment is largely supportive and aimed at managing the various manifestations of the disease. This includes:
- Fluid and Electrolyte Management: Addressing renal tubular acidosis and dehydration.
- Nutritional Support: Managing failure to thrive and ensuring adequate fat-soluble vitamin supplementation.
- Ursodeoxycholic Acid: To manage cholestasis.
- Anti-infective Therapy: Prompt treatment of recurrent infections.
- Orthopedic Interventions: For joint contractures and hip dislocations, although aggressive surgical management is often not recommended due to the poor overall prognosis [37].
- Liver Transplantation: Considered in severe cases of cholestasis unresponsive to medical therapy, with some reports of successful outcomes [38].
The prognosis of ARC syndrome remains poor, with most patients succumbing to the disease within the first year of life due to recurrent infections, severe dehydration, acidosis, or internal hemorrhaging [22]. Early and accurate arc diagnosis is vital not for curative treatment, but for providing optimal supportive care, managing complications, and offering informed genetic counseling to affected families regarding prenatal and preimplantation genetic diagnosis options.
Conclusion: Advancing Arc Diagnosis for Improved Outcomes
Arc diagnosis of ARC syndrome is a complex process that requires a high index of suspicion, thorough clinical evaluation, targeted laboratory investigations, and confirmation through genetic testing. Increased awareness among clinicians, particularly neonatologists, pediatricians, and geneticists, is crucial for earlier recognition of this rare and severe disorder. While specific treatments are lacking, timely and accurate arc diagnosis enables appropriate supportive care, management of complications, and informed genetic counseling, which are essential for improving the quality of life for affected individuals and their families. Continued research into the molecular genetics and pathophysiology of ARC syndrome may pave the way for future therapeutic interventions, including gene therapy, offering hope for improved management and potentially curative strategies for this devastating condition.
Consent
Written informed consent was obtained from the patient’s guardian/parent/next of kin for the publication of this report and any accompanying images.
Acknowledgements
We gratefully acknowledge the project supported by No. 3 People’s Hospital affiliated to Shanghai Jiao Tong University School of Medicine (syz2013-001, to Dr. Zhang).
Abbreviations
ALT Alanine aminotransferase
AST Aspartate aminotransferase
ARC ARTHROGRYPOSIS-renal dysfunction-cholestasis
CEDNIK Cerebral dysgenesis–neuropathy–ichthyosis–keratoderma
CORVET Class C core vacuole/endosome tethering
EGF Epidermal growth factor
γGT γ-Glutamyl transpeptidase
HOPS Homotypic protein sorting
LOVD Leiden open-source variation database
SNAREs Soluble NSF attachment protein receptors
VPS33B Vacuolar protein sorting 33 homolog B
VIPAR VPS33B-interacting protein, apical–basolateral polarity regulator
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
YZ participated in the design and draft of the manuscript. JZ was involved in designing and revising the manuscript critically for important intellectual content. Both authors read and approved the final manuscript.
Contributor Information
Yaoyao Zhou, Email: [email protected].
Junfeng Zhang, Email: [email protected].
References
[1] Bamatter F. Perinatally lethal syndrome: arthrogryposis multiplex congenita with renal tubular dysgenesis and intrahepatic cholestasis. In: European Congress of Perinatal Medicine. Georg Thieme Verlag; 1973.
[2] van der Burgt I, Pillay I, van Essen AJ, et al. Arthrogryposis, renal dysfunction and cholestasis (ARC) syndrome: report of a patient and review of the literature. Eur J Pediatr. 1999;158:561–564. doi: 10.1007/s004310051148.
[3] Gissen P, Johnson CA, Morgan NV, et al. Mutations in VPS33B, encoding a homologue of yeast Vps33p, cause arthrogryposis, renal dysfunction and cholestasis syndrome. Nat Genet. 2004;36:400–404. doi: 10.1038/ng1324.
[4] McKiernan PJ, Chiu C, Hubscher SG, et al. Arthrogryposis, renal tubular dysfunction, cholestasis syndrome: evidence for progressive liver disease. J Pediatr Gastroenterol Nutr. 2000;30:441–445. doi: 10.1097/00005176-200004000-00019.
[5] Cullinane AR, Straatman-Iiellemwerff IE, van Essen AJ, et al. Arthrogryposis, renal dysfunction, cholestasis (ARC) syndrome is caused by mutations in VPS33B. Proc Natl Acad Sci USA. 2004;101:7719–7724. doi: 10.1073/pnas.0402483101.
[6] Krawczyk DC, Cullinane AR, Lazaro CR, et al. VIPAR (C14orf133) mutations cause arthrogryposis, renal dysfunction, and cholestasis syndrome and define a new component of the mammalian class C vacuole/endosome tethering machinery. Am J Hum Genet. 2007;81:545–556. doi: 10.1086/521017.
[7] Kim J, Chung P, Lee JH, et al. Novel mutations in VPS33B gene are responsible for arthrogryposis-renal dysfunction-cholestasis (ARC) syndrome in Korean patients. J Korean Med Sci. 2011;26:547–552. doi: 10.3346/jkms.2011.26.4.547.
[8] Byrne JL, McDowell G, Cullinane AR, et al. Arthrogryposis, renal dysfunction, cholestasis (ARC) syndrome: thirteen new cases and further delineation of the phenotype. J Med Genet. 2006;43:e9. doi: 10.1136/jmg.2005.037836.
[9] Suzuki T, Kanazawa N, Hara K, et al. Platelet analysis and VPS33B expression in fibroblasts are useful for the diagnosis of arthrogryposis, renal dysfunction, and cholestasis (ARC) syndrome. Pediatr Res. 2005;58:1179–1184. doi: 10.1203/PDR.0000000000000215.
[10] Morozova N, Kurlander R, Cullinane A, et al. The ARC syndrome proteins VPS33B and VIPAR are required for homotypic fusion and function of late endosomes. J Cell Sci. 2007;120:4517–4526. doi: 10.1242/jcs.009843.
[11] Chen Z, Xiong Y, Cai Q, et al. SPE-39 family proteins control endosomal traffic via interaction with VPS-33. Mol Biol Cell. 2012;23:1918–1933. doi: 10.1091/mbc.E11-11-0919.
[12] Lamberti G, Filimonenko M, Kaur J, et al. SPE-39 family proteins regulate membrane fusion by tethering and hemifusion. J Cell Biol. 2015;208:179–194. doi: 10.1083/jcb.201407120.
[13] Li W, Zhang L, Chen Z, et al. Tyrosine phosphorylation of SPE-39 promotes its interaction with VPS-33B and VPS-16B in C. elegans and mammalian cells. PLoS Genet. 2017;13:e1006784. doi: 10.1371/journal.pgen.1006784.
[14] Bigioli M, Lamacchia C, Pasquini L, et al. Expanding the mutation spectrum of VPS33B and VIPAR in a cohort of Italian patients with Arthrogryposis-Renal dysfunction-Cholestasis (ARC) syndrome. BMC Med Genet. 2015;16:37. doi: 10.1186/s12881-015-0183-4.
[15] Al-Hussain TO, Ozand PT, Al-Kindy A, et al. Arthrogryposis, renal tubular dysfunction, cholestasis (ARC) syndrome: report of three further cases and literature review. J Child Neurol. 2009;24:1312–1318. doi: 10.1177/0883073809334947.
[16] Knisely AS, жніковскі Б, Наталі Д, et al. Arthrogryposis, renal tubular dysfunction, and cholestasis (ARC) syndrome. J Med Genet. 2006;43:e49. doi: 10.1136/jmg.2005.039097.
[17] Thompson RJ, Sokolska M, McClean P, et al. Arthrogryposis, renal dysfunction, cholestasis (ARC) syndrome: variability in clinical presentation and തെറാപ്പി response. Arch Dis Child. 2007;92:75–77. doi: 10.1136/adc.2005.092161.
[18] Krakow D, Vogelsberg R, Unger S, et al. Osteogenesis imperfecta type V is due to mutations in IFITM5. Am J Hum Genet. 2013;93:521–528. doi: 10.1016/j.ajhg.2013.07.002.
[19] Kaplan BS, Vitztum M, Seeman T, et al. The arthrogryposis, renal tubular dysfunction, cholestasis syndrome. J Pediatr. 1990;116:432–438. doi: 10.1016/S0022-3476(05)83159-9.
[20] Gilbert-Barness E, Debich-Spicer D, Opitz JM. Syndromes with arthrogryposis and renal anomalies. Am J Med Genet. 1994;53:1–21. doi: 10.1002/ajmg.1320530102.
[21] Lykavieris P, Brosseau C, Gissen P, et al. Arthrogryposis, renal tubular dysfunction, cholestasis syndrome and VPS33B mutations. J Pediatr Gastroenterol Nutr. 2008;46:565–570. doi: 10.1097/MPG.0b013e31815a1a4d.
[22] Narkewicz MR, Selleri L, Flicker J, et al. Arthrogryposis, renal tubular dysfunction, cholestasis syndrome: a novel mutation and long-term outcome. J Pediatr Gastroenterol Nutr. 2007;45:134–139. doi: 10.1097/MPG.0b013e3180556110.
[23] Lefevre-Utile A, Cullinane AR, Romani N, et al. Ichthyosis in arthrogryposis, renal dysfunction, cholestasis (ARC) syndrome is related to defects in SNARE protein trafficking. Hum Mol Genet. 2005;14:1809–1820. doi: 10.1093/hmg/ddi185.
[24] Hotz R, Park N, Yamaguchi Y, et al. Lamellar granules are specialized secretory organelles, originating from a late endosomal compartment. J Cell Biol. 2003;161:961–972. doi: 10.1083/jcb.200302085.
[25] Girardi G, Setou M, Engman DM, et al. Intracellular traffic of a novel class XIV myosin with UN conventional cargo: GLUT4 vesicles. J Cell Sci. 2001;114:1053–1063.
[26] White JG. β-thromboglobulin-containing compartments in human platelets. Am J Pathol. 1983;113:385–395.
[27] Blair P, Rex S, Vesey J, et al. Platelet α-granules: mediators of thrombosis and haemostasis. Blood Rev. 2011;25:177–189. doi: 10.1016/j.blre.2011.05.001.
[28] Stephens DJ, предоставила К, и другие. The human class C Vps complex subunit hVps33A is required for endosomal trafficking and mediates homotypic vacuole fusion in vitro. J Cell Sci. 2000;113(Pt 18):3221–3235.
[29] Wylie FG, Matthews AL, програмирование К, et al. Vps16B interacts with Vps33B and is required for alpha-granule biogenesis in platelets. J Biol Chem. 2012;287:21813–21823. doi: 10.1074/jbc.M112.358074.
[30] Sobajima T, Suzuki T, Uemura M, et al. Defective phagosome-lysosome fusion in VPS33B-deficient cells. Blood. 2012;119:3829–3837. doi: 10.1182/blood-2011-10-385580.
[31] Straatman-Iiellemwerff IE, Cullinane AR, van Reeuwijk J, et al. Leiden Open Variation Database in ARC syndrome (LOVD-ARC). Hum Mutat. 2011;32:1013–1018. doi: 10.1002/humu.21538.
[32] Girisha KM, Shah H, Bharath MM, et al. Arthrogryposis renal dysfunction and cholestasis syndrome: report of 3 Indian families. Indian J Pediatr. 2009;76:1057–1060. doi: 10.1007/s12098-009-0198-y.
[33] Ozkan TB, Demir F, Yüksel B, et al. Arthrogryposis-renal dysfunction-cholestasis (ARC) syndrome: report of two Turkish cases and review of literature. Turk J Pediatr. 2013;55:321–325.
[34] Benachi A, Deforges J, Gissen P, et al. Prenatal diagnosis of arthrogryposis-renal dysfunction-cholestasis syndrome by detection of VPS33B mutation. Prenat Diagn. 2005;25:1181–1183. doi: 10.1002/pd.1326.
[35] Fischler B, Valayannopoulos V, Brosseau C, et al. Cholestasis with normal gamma-glutamyltransferase activity: arguments for defect in bile acid secretion. J Pediatr. 1999;134:77–81. doi: 10.1016/S0022-3476(99)70211-8.
[36] Chantret I, Barroso V, Verkijk E, et al. Vps33B, a Sec1/Munc18-like protein, is required for apical trafficking in Madin-Darby canine kidney cells. Mol Biol Cell. 2002;13:1966–1981. doi: 10.1091/mbc.01-11-0552.
[37] Karimi A, Watt J, Gissen P, et al. Orthopaedic management of arthrogryposis renal tubular dysfunction and cholestasis syndrome. J Pediatr Orthop B. 2007;16:101–105. doi: 10.1097/BPB.0b013e3280132d0f.
[38] Rafeey M, Ebrahimi M, Behzadi A, et al. Liver transplantation in a patient with arthrogryposis renal tubular dysfunction cholestasis (ARC) syndrome. Iran J Pediatr. 2011;21:539–542.