Introduction
Autoimmune atrophic gastritis (AAG) is a chronic autoimmune condition targeting the oxyntic mucosa of the stomach. This immune-mediated destruction is primarily driven by anti-parietal cell antibodies and/or anti-intrinsic factor antibodies. While the exact mechanisms of AAG pathogenesis are still under investigation, a T helper cell type 1 (Th1) response appears to be central, with autoantibodies potentially arising as a consequence of cellular damage to the H+/K+ ATPase pump [1,2]. This inflammatory process leads to the loss of parietal cells, which are crucial for gastric acid and intrinsic factor production. Notably, AAG preferentially affects the gastric body and fundus, typically sparing the antrum [1,3]. This distribution contrasts with Helicobacter pylori (H. pylori) gastritis, which commonly involves the antrum and can extend to the corpus, resulting in pangastritis. The interplay between H. pylori infection and AAG remains a complex area of research, with ongoing debate about whether they represent distinct entities [4].
Autoimmune gastritis is increasingly recognized as a significant clinical entity due to its potential to cause malabsorption, vitamin deficiencies, and elevate the risk of gastric malignancies, including neuroendocrine tumors and adenocarcinoma. Accurate and timely Autoimmune Atrophic Gastritis Diagnosis is therefore paramount for effective patient management and risk mitigation.
This review provides an updated and comprehensive guide to AAG, with a particular focus on autoimmune atrophic gastritis diagnosis. We will explore recent evidence concerning epidemiology, pathogenesis, clinical presentation, malignancy risks, and current diagnostic and management strategies.
Epidemiology
Determining the precise prevalence of AAG in the general population is challenging due to the absence of universally standardized diagnostic criteria. Diagnostic approaches vary across studies, with some relying on histological examination of gastric biopsies, while others use surrogate markers like low vitamin B12 levels or serological tests for anti-parietal cell and anti-intrinsic factor antibodies.
Current estimates suggest that AAG prevalence ranges from 0.3% to 2.7% in the general population [5]. Geographical and ethnic variations in AAG prevalence have been observed, with higher rates reported in Western countries compared to Eastern countries [1]. However, this disparity may be partially attributed to a higher incidence of H. pylori-related atrophic gastritis in Asian populations, potentially leading to misdiagnosis and underestimation of AAG. A study in Japan highlighted the potential for underdiagnosis of AAG, particularly after H. pylori eradication, suggesting that the true prevalence may be higher than currently recognized [6].
AAG is more frequently diagnosed in women, older individuals (over 60 years), and those with pre-existing autoimmune disorders. The female-to-male ratio is approximately 2–3:1 [7,8]. Individuals with a personal or family history of autoimmune conditions, notably type 1 diabetes and autoimmune thyroiditis, have a significantly elevated risk of AAG, with a prevalence five times greater than in control populations [9]. In fact, AAG appears to be the most common autoimmune comorbidity in patients with autoimmune thyroiditis, with a reported prevalence of 2.8% [10]. Conversely, a retrospective study found that autoimmune thyroiditis was present in 36% of AAG patients, followed by rheumatoid arthritis (9%), systemic lupus erythematosus (6%), and celiac disease (3%) [11,12].
Etiopathogenesis
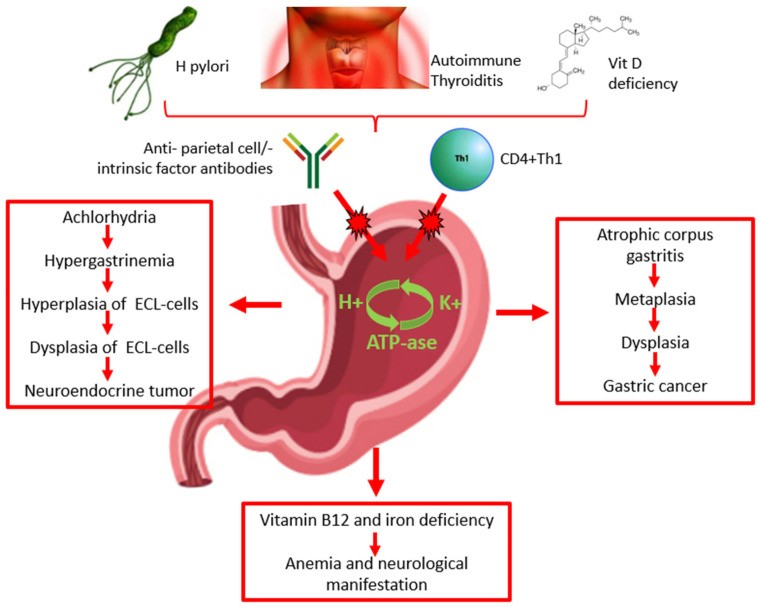
The precise mechanisms driving AAG pathogenesis are still being elucidated. A key feature is chronic gastric mucosal inflammation, often associated with anti-parietal cell antibodies and, less frequently, anti-intrinsic factor antibodies. These autoantibodies are detectable in the serum of 60–90% and 50–70% of AAG patients, respectively (Figure 1) [13,14]. However, some evidence suggests that anti-parietal cell antibodies may not be the primary pathogenic factor. Instead, they might be a secondary consequence of T cell-mediated damage to the H+/K+ ATPase pump, leading to the exposure of self-antigens and subsequent autoantibody production (Figure 1) [1,2]. Emerging research indicates a potential role for vitamin D deficiency in the pathogenesis of autoimmune diseases, including AAG. Vitamin D receptors are involved in T-cell activation and differentiation, crucial for maintaining immune tolerance and self-antigen recognition. Vitamin D deficiency could therefore disrupt T-cell function and increase susceptibility to autoimmune disorders like AAG (Figure 1) [15].
Figure 1.
Pathogenesis, diagnosis, clinical presentation of autoimmune atrophic gastritis, and risk of malignancy. ECL: enterochromaffin-like.
The relationship between H. pylori infection and AAG is complex and not fully understood. H. pylori may contribute to AAG induction or exacerbation (Figure 1). Patients with H. pylori-positive corpus atrophic gastritis often exhibit elevated anti-parietal cell antibody levels, which tend to decrease after H. pylori eradication. Parietal cell damage from H. pylori-related inflammation could expose ATPase pump antigens, triggering anti-parietal cell antibody production through molecular mimicry between bacterial and self-antigens. This is more likely to occur in genetically predisposed individuals with specific MHC class II haplotypes [16] (Table 1). Case reports suggest that H. pylori-related gastritis can transition into AAG, and the progression of AAG may even lead to spontaneous H. pylori clearance due to worsening gastric atrophy [17].
Table 1.
Recent findings in the topic of atrophic autoimmune gastritis.
| References | Finding |
|---|---|
| Iwamuro M, Curr Issues Mol Biol 2023 [16] | Helicobacter pylori may play a role in the induction/exacerbation of AAG. |
| Miceli E, Am J Gastroenterol 2023 [20] | The annual rate of progression of 10.9% from “potential” to “overt” AAG would suggest that anti-parietal cell antibodies are a true marker of “potential” AAG in patients without corpus atrophy. |
| Guo X, J Clin Med 2023 [21] | Anti-parietal cell antibodies, serum gastrin, PGI/PGII ratio, and vitamin B12 can be useful to identify patients with AAG among those with precancerous conditions, i.e., atrophic gastritis and intestinal metaplasia. |
| Singh S, Cureus 2023 [22] | Patients with AAG are often asymptomatic, but they can refer dyspeptic or reflux symptoms. |
| Conti L, Microorganisms 2020 [23] | Hypochloridria results in alteration of composition of gastric microbiota and patients with AAG have higher microbial diversity. |
| Miceli E, Dig Liv Dis 2023 [24] | Atrophic autoimmune gastritis can be linked to infertility, recurrent miscarriage, congenital abnormalities, and several obstetric complications. |
| Rugge M, GUT 2023 [25] | In patients with AAG, the cumulative incidence of type 1 neuroendocrine tumors (NETs) is 4.7% at 2 years of follow-up. |
| Dilaghi E, Am J Gastroenterol 2023 [26] | The incidence rate of gastric cancer/high grade dysplasia is 0.5% per person/year; risk factors for gastric cancer are age >60 years, intestinal metaplasia with absence of pseudopyloric metaplasia, pernicious anemia. |
AAG: atrophic autoimmune gastritis.
Conversely, murine model studies suggest that H. pylori infection might suppress AAG development. AAG is driven by a CD4+ Th1 response, which appears to be downregulated in H. pylori-infected mice due to Th2-type immune responses and transforming growth factor β [18]. This aligns with a case report of a woman who developed AAG after H. pylori eradication, with rapid progression of corpus atrophy within three years. The authors proposed that H. pylori gastritis might have masked AAG activity until eradication [19]. The low AAG prevalence in Asian countries, where H. pylori infection is highly prevalent, also supports this complex interaction [18].
AAG development likely involves a combination of genetic and environmental factors. A strong genetic predisposition is evident in individuals with a family history of autoimmune disorders. The association between AAG and other autoimmune conditions underscores the role of genetic susceptibility, particularly HLA haplotypes [10]. The pathogenic link between autoimmune thyroiditis and AAG is still under investigation. One prominent hypothesis suggests immunological cross-reactivity, with a shared molecular pattern identified between thyroid peroxidase and the H+/K+ ATPase of gastric parietal cells (Figure 1) [27].
Further research has implicated chemokine receptor type 7 (CCR7) deficiency in AAG development in mice [28]. CCR7 is crucial for B and T lymphocyte activation and dendritic cell maturation, processes essential for maintaining immune tolerance and self-antigen recognition [29].
4. Autoimmune Atrophic Gastritis Diagnosis
Autoimmune atrophic gastritis diagnosis relies on a combination of serological markers and histological evaluation of gastric biopsies (Figure 1). A comprehensive diagnostic approach is crucial for accurate identification and appropriate management of AAG.
4.1 Serological Diagnosis
Serological testing plays a significant role in autoimmune atrophic gastritis diagnosis. Key markers include anti-parietal cell antibodies and anti-intrinsic factor antibodies [30].
-
Anti-parietal cell antibodies (APCA): These antibodies exhibit high sensitivity (81%) and specificity (90%) for AAG diagnosis [1]. While a positive predictive value is relatively low (around 20% based on a 3% AAG prevalence), the negative predictive value is excellent (99%). This makes APCA testing a highly effective tool for ruling out AAG. However, it’s important to note that APCA can be present years before overt AAG develops, and not all APCA-positive individuals will develop histological AAG. APCA titers may also decline with age, potentially reducing their reliability in elderly patients [5].
-
Anti-intrinsic factor antibodies (AIFA): AIFA have lower sensitivity (27%) but very high specificity (100%) for AAG diagnosis [1]. Due to their low sensitivity, AIFA are less useful as a primary screening tool but can be highly confirmatory when positive, especially in cases of suspected pernicious anemia.
The concept of “potential AAG” has emerged, analogous to celiac disease. Potential AAG is defined by the presence of APCA in the absence of gastric atrophy (at any site) and without active H. pylori infection. Approximately 50% of individuals with potential AAG progress to overt AAG within a median of two years [31] (Table 1). A recent prospective study reported an annual progression rate of 10.9% from potential to overt AAG, reinforcing APCA as a marker for potential AAG in the absence of corpus atrophy [20] (Table 1). Early histopathological markers of potential AAG are still under investigation, with increased CD3+ intraepithelial lymphocytes being a potential early finding [32]. Predictors of progression from potential to overt AAG are still being researched.
Serum pepsinogens I and II, and gastrin-17 levels are also valuable in AAG diagnosis.
- Pepsinogen I (PG-I): Produced by the oxyntic mucosa, PG-I serum levels are typically low in AAG due to oxyntic atrophy [33].
- Pepsinogen II (PG-II): Produced by both oxyntic and antral mucosa, PG-II levels may be less affected by AAG.
- PG-I/PG-II Ratio: A low PG-I/PG-II ratio is a sensitive indicator of corpus atrophic gastritis.
- Gastrin-17: Synthesized by antral G cells, serum gastrin-17 levels are often elevated in AAG due to hypo- or achlorhydria resulting from oxyntic atrophy [34].
Combining these serum tests forms the basis of the Gastropanel®, a commercially available diagnostic tool that assesses serum PG-I, PG-II, gastrin-17, and anti-Helicobacter pylori antibodies. Low PG-I or PG-I/PG-II ratio combined with high gastrin-17 levels strongly suggest corpus atrophic gastritis. Meta-analysis of studies evaluating Gastropanel® performance for corpus-limited atrophic gastritis reported a summary sensitivity of 70.4% and specificity of 98.4% [35].
Serum chromogranin A levels are often elevated in AAG patients, but this marker lacks specificity and is not recommended as a primary diagnostic test for AAG [36].
4.2 Endoscopic Diagnosis
Endoscopy with careful mucosal inspection is crucial in the autoimmune atrophic gastritis diagnosis process. Endoscopic findings in AAG can include mucosal erythema, nodularity, and a hypo-atrophic mucosal pattern. However, white-light endoscopy alone has limitations in detecting subtle mucosal atrophy.
Advanced endoscopic techniques enhance diagnostic accuracy:
- High-definition (HD) endoscopy: Provides improved mucosal visualization.
- Chromoendoscopy: Dye-based or virtual (narrow-band imaging – NBI) chromoendoscopy highlights mucosal surface details and vascular patterns. In normal oxyntic mucosa, a honeycomb subepithelial capillary network and regular venous collection are visible, which are often absent in atrophic gastritis [37].
- Magnification endoscopy: Allows for closer examination of mucosal microstructure.
- Autofluorescence endoscopy: Detects subtle mucosal changes based on tissue autofluorescence properties.
While advanced endoscopy improves detection of mucosal abnormalities, histological confirmation remains essential for definitive autoimmune atrophic gastritis diagnosis.
4.3 Histopathological Diagnosis
Histological evaluation of gastric biopsies is the gold standard for autoimmune atrophic gastritis diagnosis [8]. Current guidelines (MAPS II) recommend obtaining at least two biopsies from the antrum and two from the corpus, collected in separate containers [38]. The Updated Sydney System also recommends an additional biopsy from the incisura angularis [39].
The hallmark of histological autoimmune atrophic gastritis diagnosis is atrophy confined to the corpus mucosa. Histologically, AAG progresses through stages:
- Early phase: Lamina propria inflammation predominantly with CD4+ lymphocytes, variable pseudopyloric or pancreatic acinar metaplasia, mild to moderate atrophy, and potentially hypertrophic residual parietal cells due to hypergastrinemia.
- Florid phase: Moderate to severe oxyntic gland atrophy with lymphoplasmacytic inflammation, enterochromaffin-like (ECL) cell hyperplasia, and predominantly intestinal metaplasia in the oxyntic mucosa. Antral biopsies may show gastrin cell hyperplasia (Figure 2).
- End phase: Severe oxyntic gland loss, intestinal metaplasia, ECL cell hyperplasia (linear or nodular), and reduced mucosal inflammation. ECL cell hyperplasia is a precursor to type 1 neuroendocrine tumors [2].
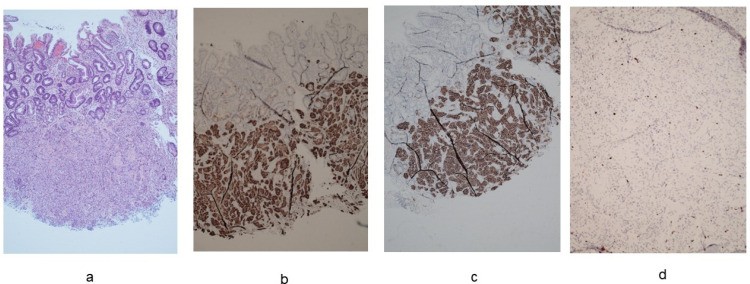
Figure 2.
Histological features of autoimmune atrophic gastritis. (a). Oxyntic mucosa with diffuse pseudopyloric and intestinal metaplasia (hematoxylin-eosin staining) (10× magnification). (b). Intraglandular linear and nodular hyperplasia of the enterochromaffin-like cells (chromogranin A staining) (20× magnification). (c). Hyperplasia of gastrin G cells in antrum (hematoxylin-eosin staining) (20× magnification).
AAG is often underdiagnosed in patients with precancerous conditions like atrophic gastritis and intestinal metaplasia in the antrum and/or corpus. Studies show a higher prevalence of AAG in patients with precancerous conditions compared to controls [21]. In patients with atrophic gastritis and intestinal metaplasia, utilizing APCA testing, serum gastrin, PG-I/PG-II ratio, and vitamin B12 levels can aid in identifying underlying AAG (Table 1) [21].
Given the association between AAG and other autoimmune diseases, screening for thyroid disorders or type 1 diabetes is advisable in patients with confirmed AAG. Conversely, screening for AAG in patients with autoimmune thyroiditis can improve the detection rate of occult gastric atrophy [40].
5. Clinical Manifestations
Patients with AAG can be asymptomatic, making autoimmune atrophic gastritis diagnosis challenging in early stages. However, many patients experience dyspeptic or reflux-like symptoms (Table 1) [22]. Symptoms may be intermittent or persistent and can be triggered or exacerbated by certain foods or fasting. A study of 379 AAG patients found that 57% reported dyspeptic symptoms, more common in younger individuals, women, non-smokers, and those without pernicious anemia [41]. Dyspepsia in AAG may be related to hypochlorhydria and hypergastrinemia, potentially delaying gastric emptying and causing postprandial fullness or early satiety [42,43]. Some AAG patients report heartburn and regurgitation [44]. However, impedance-pH monitoring often does not reveal abnormal acid reflux, suggesting that these symptoms might be due to non-acid reflux or functional mechanisms [45].
Anemia, particularly pernicious anemia and iron deficiency anemia, are common complications of AAG (Figure 1). Pernicious anemia results from vitamin B12 (cobalamin) deficiency, leading to macrocytic anemia. Vitamin B12 malabsorption is a direct consequence of oxyntic atrophy, as parietal cells produce intrinsic factor, essential for vitamin B12 absorption in the terminal ileum [47,48,49]. Iron deficiency anemia is also frequent in AAG and may precede pernicious anemia. Hypochlorhydria in AAG impairs the reduction of dietary iron from Fe3+ to the absorbable Fe2+ form [23].
Vitamin B12 deficiency can also cause fatigue, weakness, and neurological complications. Cobalamin is a coenzyme in fatty acid metabolism, and deficiency can lead to nerve demyelination and axonal damage, resulting in peripheral neuropathies ranging from paresthesia to paraparesis (Figure 1) [50].
A large study of 168 women suggested a link between AAG and infertility, recurrent miscarriage, congenital abnormalities, and obstetric complications [24]. Vitamin B12 deficiency is known to affect pregnancy outcomes, but this study highlights a potential broader association of AAG with pregnancy complications. AAG should be considered as a potential comorbidity in women with recurrent pregnancy loss, particularly in the context of polyautoimmunity and after excluding antiphospholipid syndrome [51]. Therefore, AAG diagnosis may be relevant for women seeking pregnancy (Table 1) [24].
6. Risk of Malignancy
Autoimmune atrophic gastritis is a preneoplastic condition associated with increased risk of type I neuroendocrine tumors (NETs) and gastric adenocarcinoma [52] (Figure 1).
Type I NETs (carcinoids) arise from ECL cells (Figure 3). Parietal cell loss and achlorhydria lead to hypergastrinemia, stimulating ECL cell hyperplasia and potentially progressing to NETs [36,53]. Gastric body glandular polyps in AAG patients are strongly associated with type I carcinoids [54]. The odds ratio for developing type I NETs in AAG is approximately 11 [52]. Recent cohort studies report varying incidence rates of type I NETs in AAG patients, ranging from 2.8% to 4.8% per person-year [20,25,26] (Table 1). However, type I NETs generally have a favorable prognosis with low metastatic potential [55].
Figure 3.
Neuroendocrine tumor (NET) G1, diameter: 3 mm, ki-67: 1.4%. (a). Hematoxylin-eosin staining (20× magnification). (b). Immunohistochemical staining for synaptophysin (20× magnification). (c). Immunohistochemical staining for SSTR2 (20× magnification). (d). Immunohistochemical staining for ki67 (40× magnification).
The association between AAG and gastric adenocarcinoma risk is less clear [56]. Some studies suggest a threefold increased risk of gastric adenocarcinoma in AAG patients compared to the general population [55,57]. However, many studies focused on patients with pernicious anemia (severe AAG), potentially introducing selection bias. One cohort study reported a gastric cancer/high-grade dysplasia incidence rate of 0.5% per person-year in AAG, with risk factors including age >60 years, intestinal metaplasia without pseudopyloric metaplasia, and pernicious anemia [26] (Table 1).
Gastric adenocarcinoma development follows the Correa cascade: inflammation, metaplasia, atrophy, dysplasia, and carcinoma [58]. Severe atrophy carries a higher gastric cancer risk compared to mild atrophy [55]. Hypochlorhydria in AAG alters gastric microbiota [59], with increased microbial diversity and Streptococci abundance [60,23], bacteria also found in gastric cancer. However, the link between microbiota changes and gastric cancer in AAG requires further investigation.
Conversely, some studies suggest AAG may not significantly increase gastric adenocarcinoma risk, particularly in H. pylori-negative patients. One study found no invasive gastric cancers (excluding NETs) in AAG patients followed for 7.5 years [25]. They noted that metaplasia in AAG is often pseudopyloric or complete intestinal metaplasia, while incomplete intestinal metaplasia, considered higher risk for adenocarcinoma progression, is less common. They hypothesize that AAG, in the absence of incomplete intestinal metaplasia, may not predispose to adenocarcinoma, and prior H. pylori infection might explain reported gastric cancer risks in AAG. Another study reported no gastric adenocarcinoma cases in AAG patients followed for a median of 52 months, with neoplastic complications primarily being NETs and epithelial dysplasia [20]. Murine models suggest that M2-macrophages, pro-carcinogenic immune cells, are less prevalent in AAG without H. pylori compared to H. pylori-related gastritis [61,62]. This difference in macrophage infiltration may contribute to a more benign metaplastic pattern in AAG and potentially explain the debated adenocarcinoma risk [63].
7. Management
AAG management focuses on addressing vitamin and iron deficiencies and early detection of pre-neoplastic and neoplastic lesions through endoscopic surveillance. No specific treatment directly targets the autoimmune process in AAG. Systemic corticosteroids are not suitable for long-term use due to side effects. Research is ongoing into locally acting anti-inflammatory agents to reduce gastric inflammation and prevent atrophy and metaplasia progression.
Proton pump inhibitors (PPIs) may not be beneficial and could worsen nutrient malabsorption in AAG. H2 receptor antagonists (e.g., famotidine) might provide symptom relief for heartburn and epigastric burning with less impact on iron absorption. Sucralfate and other mucosal protectants may promote mucosal healing by forming a protective barrier [64].
Ghrelin, a hormone regulating appetite and gastric motility, is being investigated as a potential therapy to improve upper gastrointestinal symptoms in AAG [65]. Fecal microbiota transplantation and prebiotic use are also being explored to restore gastric microbiota balance in AAG patients with altered gastric environments due to hypochlorhydria [66].
Vitamin B12 deficiency is effectively treated with intramuscular or sublingual vitamin B12 supplementation. Prompt treatment is crucial to prevent irreversible neurological damage [67].
Endoscopic surveillance is essential for malignancy risk management. ESGE guidelines (MAPS II) recommend endoscopic surveillance with antral and corpus biopsies every 3 to 5 years in AAG patients [38]. High-definition endoscopy with chromoendoscopy (dye-based or virtual, like NBI) is recommended for high-quality mucosal assessment [68]. Visible lesions should be classified using the Paris classification [69]. Non-invasive gastric cancer detection methods using non-coding RNA are under investigation but not yet in clinical practice [70].
ESGE guidelines recommend endoscopic resection for type 1 NETs larger than 10 mm [71]. Endoscopic ultrasound can assess tumor invasion and lymph node metastasis in larger tumors. Post-resection endoscopic follow-up is suggested every 6 to 12 months [71].
Medical treatment of NETs with the gastrin-receptor antagonist netazepide has shown promise in reducing serum chromogranin A levels and potentially tumor size [72,73]. Somatostatin analogues are another option. One study showed somatostatin analogues reduced gastrin and chromogranin A levels and led to carcinoid disappearance in some patients after a median treatment duration of 12 months [74]. However, these treatments require continuous administration as tumors may regrow upon cessation [72,74].
8. Conclusions
Autoimmune atrophic gastritis diagnosis remains a crucial clinical challenge, as the condition is frequently underdiagnosed, particularly in subclinical forms. The epidemiology of AAG requires further clarification due to the lack of standardized diagnostic criteria, potentially leading to underestimation of its prevalence. The etiopathogenesis of AAG is complex and not fully understood, necessitating ongoing research.
The concept of “potential AAG,” defined by APCA positivity without histological atrophy or H. pylori infection, is a significant development in understanding the early stages of the disease. Further research is needed to elucidate the natural history of AAG and identify risk factors for disease progression. While the association between AAG and NETs is well-established, the risk of gastric adenocarcinoma remains debated, with some evidence suggesting it may be less significant than previously thought, particularly in H. pylori-negative patients.
Management of AAG centers on correcting vitamin and iron deficiencies and implementing endoscopic surveillance for early cancer detection. Early autoimmune atrophic gastritis diagnosis is crucial to ensure timely vitamin B12 and iron supplementation, ideally before the onset of anemia and neurological symptoms. Continued research is essential to develop new therapies aimed at reducing gastric inflammation and preventing the progression of atrophy and metaplasia in AAG.
Author Contributions
C.C.: conceptualization, writing-original draft preparation, and data curation; L.H.E.: writing-original draft preparation, review, and editing; E.D.: conceptualization, writing, review, and editing; A.V.: review and editing; P.F.: review and editing; L.F.: review and editing; A.D.: review and editing; V.I.: review and editing; R.M.Z.: conceptualization, writing-original draft preparation, and review data curation. All authors have read and agreed to the published version of the manuscript.
Conflicts of Interest
The authors declare no conflicts of interest.
Funding Statement
This research received no external funding.
Footnotes
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content.
References
[1] Sugano, K. Autoimmune gastritis. Curr. Opin. Gastroenterol. 2000, 16, 475–479.
[2] Park, J.Y.; Koh, Y.W.; Kim, J.E.; Lee, H.E.; Yang, H.K.; Kim, W.H.; Kim, H.H. Carcinoids of the stomach associated with autoimmune atrophic gastritis: Clinicopathologic and immunohistochemical characteristics. Am. J. Surg. Pathol. 2007, 31, 1560–1569.
[3] Fisker, N.O.; Fenger, C.; выпадение, L.P. Autoimmune atrophic gastritis in patients with fundic gland polyps and colorectal adenomas. APMIS 2015, 123, 520–527.
[4] Fallon, A.; Maecker, B.; Vogl, M.E.; Prenner, G.; Haybar, H.; Kiesslich, T.; Broessler, A.; Vieth, M.; Behboudi, S.; Trauner, M.; et al. Helicobacter pylori-associated pangastritis and autoimmune atrophic gastritis are distinct disease entities. Histopathology 2021, 78, 432–442.
[5] ভেগা, কে.; ক্রুজ, এম.জে.; ব্রুজো, ই.; জিউসেল, জে.এ.; কার্নিসের, জে.; গুইলারম্যাট, ই.; সেরিওল, এফ.; আরদিজোন, জে.; ভেরগেজ, জে.পি.; ভেগা, পি.; এট আল। প্রিভ্যালেন্স অফ অটোইমিউন গ্যাস্ট্রিটিস ইন জেনারেল পপুলেশন অ্যান্ড ইন সিলেক্টেড রিস্ক গ্রুপস। ইউরো। জে. গ্যাস্ট্রোএন্টারল। হেপাটোল। 2012, 24, 1173–1179.
[6] ইওয়ামুরো, এম.; আন্দো, টি.; মায়েদা, ও.; ওহমি, কে.; নাকাতা, এম.; ইশিগুরা, টি.; হিরামাতসু, এ.; মরিওয়াকি, ওয়াই.; ইগারাশি, এম.; নাগোয়া, এইচ.; এট আল। আনএক্সপেক্টেডলি হাই প্রিভ্যালেন্স অফ অটোইমিউন অ্যাট্রোফিক গ্যাস্ট্রিটিস এমং পেশেন্টস উইথ নেগেটিভ হেলিকোব্যাক্টর পাইলোরি ইনফেকশন। জোনালি অফ গ্যাস্ট্রোএন্টারোলজি অ্যান্ড হেপাটোলজি 2019, 34, 1936–1942.
[7] Koido, S.; Sugano, K. Autoimmune gastritis. জোনালি অফ গ্যাস্ট্রোএন্টারোলজি 2006, 41, 1013–1022.
[8] টোথ, ই.; ল্যাচেল, ও.; মেইজার, জে.; সিজ, এল.; বেনেদেক, জি.; রুজ, এম.; ভেজ, জে.পি.; ইয়াগুইজ, ভি.; গিয়ার্ডেলা, সি.; ওভেরাদ, টি.; এট আল। ইউরোপিয়ান প্রোটোকল ফর দ্য ডায়াগনোসিস অ্যান্ড ম্যানেজমেন্ট অফ প্রি-ক্যান্সারাস কন্ডিশনস অ্যান্ড লেশনস ইন দ্য স্টোমাক (MAPS II)। এন্ডোস্কোপি 2016, 48, 877–890.
[9] ম মুহসেন, কে.; আল-জেবুরি, এম.এম.; ইসমাইল, এস.টি.; আল-হামদানি, এন.; টমাস, জি.এন.; স্ট্র্যাডলিং, পি.এইচ.; র্যামসে, ডি.ই.; ওয়েয়ার, আর.ডি.; কেলি, ডি.ওয়াই. অটোইমিউন গ্যাস্ট্রিটিস অ্যান্ড এর এসোসিয়েশন উইথ আদার অটোইমিউন কন্ডিশনস: এ পপুলেশন-বেজড স্টাডি। জোনালি অফ অটোইমিউনিটি 2016, 71, 89–93.
[10] অ্যান্টোনেলি, এ.; ফার্সিও, এ.; মারিওত্তি, এল.; ফেরি, সি.; গালিয়াচি, জি.; ফ্যালোর্নি, এ.; সান্টোরো, এ.; মন্টি, এম.; কাসানোভা, ভি.; পারিস, এল.; এট আল। প্রিভ্যালেন্স অফ অটোইমিউন গ্যাস্ট্রিটিস ইন পেশেন্টস উইথ অটোইমিউন থাইরয়েড ডিজিজ। ইউরো। জে. এন্ডোক্রিনল। 2015, 172, 665–671.
[11] কারো, এ.সি.; রড্রিগেজ-ক্যাস্টেলান, জে.; কুইরোগা, এ.; গঞ্জালেজ, ই.; জিমেনেজ, পি.; গালাচে, সি.; ভের্গারা, পি.; ভের্গারা, এম.; ভের্গারা, এন.; কুইরোগা, জে. অটোইমিউন গ্যাস্ট্রিটিস: এ রেট্রোস্পেক্টিভ কোহর্ট স্টাডি। ইউরো। জে. গ্যাস্ট্রোএন্টারল। হেপাটোল। 2017, 29, 1418–1423.
[12] রুবিন, এম.; ডাইস, এ.; রুবিন, ডি.ই.; ফিন, ই.; শ্মিট, এ.; মিলার, এম.; টুনার, এ.; গাজো, সি.; ডেভিডসন, এম.; গাজো, সি.; এট আল। অটোইমিউন গ্যাস্ট্রিটিস ইন পেশেন্টস উইথ সেলিয়াক ডিজিজ। আম. জে. গ্যাস্ট্রোএন্টারল। 2013, 108, 1549–1555.
[13] Valako