Introduction
Beckwith-Wiedemann syndrome (BWS) is a complex imprinting disorder characterized by a spectrum of clinical features, most notably overgrowth. This condition arises from genetic and epigenetic alterations in the 11p15 chromosomal region, a critical area housing genes that regulate both prenatal and postnatal growth. Diagnosed typically in infancy or early childhood, BWS presents with varying degrees of severity. Key features include macroglossia (enlarged tongue), abdominal wall defects like omphalocele, lateralized overgrowth (asymmetrical growth of body parts), enlarged internal organs, and a heightened susceptibility to embryonal tumors in early childhood. The understanding of BWS has evolved to encompass a spectrum (BWSp), ranging from classic presentations to cases with isolated lateralized overgrowth. With an estimated incidence of 1 in 10,340 live births, BWS prevalence is noted to be higher in pregnancies conceived through assisted reproductive technologies (ART), approximating 1 in 1,100.
Clinical diagnosis of BWS and the decision to pursue molecular testing rely on a clinical scoring system. This system assigns points to both cardinal and suggestive clinical features. Cardinal features, each worth 2 points, include macroglossia, omphalocele, lateralized overgrowth, bilateral Wilms tumor, hyperinsulinism, and specific pathological findings (adrenal cytomegaly, placental mesenchymal dysplasia). Suggestive features, each worth 1 point, encompass high birth weight (over two standard deviations above the mean), facial nevus simplex, polyhydramnios or placentomegaly, ear creases or pits, transient hypoglycemia, embryonal tumors, nephromegaly or hepatomegaly, and umbilical hernia or diastasis recti. A clinical score of ≥2 warrants genetic testing for BWS. A score of ≥4, based on both cardinal and suggestive features, is sufficient for a clinical diagnosis of classical BWS, even without molecular confirmation. Scores between 1 and 3 are considered indicative of BWSp, necessitating close monitoring and further evaluation.
Mosaicism, a condition where different cells within an individual have varying genetic or epigenetic compositions, is a notable characteristic of BWS. This mosaic nature implies that genetic testing might need to be performed on multiple tissue types, as the proportion of cells carrying the BWS-related genetic change can vary across the body. Consequently, a negative genetic test result does not definitively rule out BWS, especially due to potential tissue mosaicism. Further details on molecular genetic testing for BWS are discussed below.
Molecular Mechanisms Underlying Beckwith-Wiedemann Syndrome
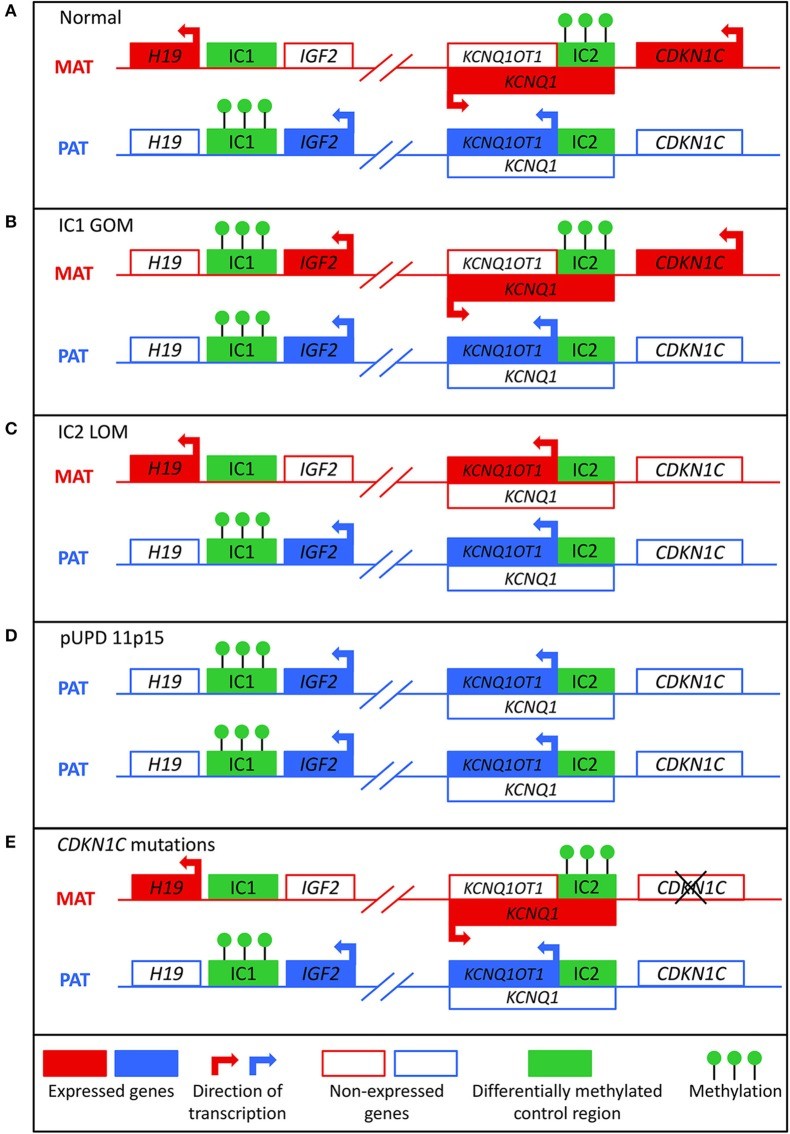
BWS is rooted in molecular anomalies within a cluster of imprinted genes located on chromosome 11p15.5-11p15.4, as illustrated in Figure 1. This region is organized into two functionally distinct domains: telomeric and centromeric, each regulated by its own imprinting control region. The telomeric domain includes the H19 long non-coding RNA (lncRNA) gene and the insulin-like growth factor 2 (IGF2) gene. H19, typically expressed from the maternal allele in the embryo and placenta, is silenced in most postnatal tissues except cardiac and skeletal muscle and is implicated in both tumor development and suppression. IGF2, a paternally expressed growth factor in the fetus and placenta, shows biallelic expression in the liver postnatally. The imprinting of the telomeric domain is governed by the H19/IGF2 intergenic differentially methylated region (H19/IGF2:IG DMR), also known as imprinting control region 1 (IC1), located upstream of H19. CTCF binding factor-dependent insulators and shared enhancers between H19 and IGF2 regulate imprinting at this locus. In a normal scenario, CTCF binds to the ICR on the maternal allele, promoting H19 expression and Igf2 silencing, while methylation of the ICR on the paternal allele prevents CTCF binding, leading to Igf2 expression and H19 silencing.
Figure 1. Molecular Mechanisms in Beckwith-Wiedemann Syndrome
Image alt text: Diagram illustrating the molecular mechanisms leading to Beckwith-Wiedemann syndrome, including normal methylation patterns and aberrations such as IC1 GOM, IC2 LOM, pUPD, and CDKN1C mutations.
The centromeric domain comprises KCNQ1, the regulatory lncRNA KCNQ1OT1, and CDKN1C, a cell cycle inhibitor. KCNQ1, encoding a voltage-gated potassium channel, is initially maternally expressed but becomes biallelically expressed during fetal development. KNCQ1OT1 is a paternally expressed lncRNA transcribed antisense to KCNQ1. CDKN1C, encoding a G1 cyclin-dependent kinase inhibitor, negatively regulates cell growth and proliferation and is expressed both pre- and postnatally. The centromeric domain is regulated by the KCNQ1OT1 transcription start site differentially methylated region (KCNQ1OT1:TSS DMR), or imprinting control region 2 (IC2), located at the 5′ end of KCNQ1OT1. Normally, the maternal allele is methylated at IC2, preventing Kcnq1ot1 expression and allowing Kcnq1 and Cdkn1c expression, while on the paternal allele, the unmethylated Kcnq1ot1 promoter permits lncRNA expression and silences Kcnq1 and Cdkn1c.
Approximately 80% of BWS patients have identifiable molecular defects in the 11p15 region, with aberrant DNA methylation being the most common. In normal cells, the paternal allele is methylated at IC1 and the maternal allele at IC2 (Figure 1A). These methylation marks, established in the germline, are typically maintained through post-fertilization reprogramming. Gain of methylation at IC1 on the maternal allele (IC1 GOM), found in 5–10% of BWS cases, leads to H19 downregulation and IGF2 overexpression from the maternal allele (Figure 1B). Loss of methylation at IC2 on the maternal allele (IC2 LOM), the most frequent molecular defect in BWS (around 50% of cases), results in KCNQ1OT1 derepression and CDKN1C downregulation from the maternal allele (Figure 1C). Paternal uniparental isodisomy (pUPD), where an individual inherits two copies of the paternal chromosome 11p15 and no maternal copy, occurs in about 20% of patients (Figure 1D). pUPD can be segmental or genome-wide, leading to IGF2 overexpression and reduced CDKN1C expression. CDKN1C mutations on the maternal allele are found in approximately 5% of sporadic BWS cases and 40% of familial cases (Figure 1E). Maternally inherited mutations carry a 50% recurrence risk and variable expressivity. Chromosomal abnormalities like duplications, deletions, and translocations of the 11p15 region account for a smaller percentage of cases. These are typically inherited in an autosomal dominant manner, with recurrence risk depending on the parent of origin of the affected allele.
Multiple gestations, particularly monozygotic female twins with discordant BWS features, are more frequent in BWS. The “diffused mosaicism” theory suggests that an epigenetic event causing BWS may precede and possibly trigger twinning, leading to a mosaic distribution of affected cells across embryos in multiple pregnancies and varying degrees of phenotypic discordance.
Assisted reproductive technologies (ART) are associated with a tenfold increased risk of BWS, with IC2 LOM being the most common molecular anomaly in ART-conceived BWS patients. Further research is needed to fully understand the link between ART and imprinting defects.
Molecular Genetic Testing Strategies for BWS Diagnosis
Overview of Diagnostic Approaches
Mosaicism significantly complicates genetic testing for BWS, as affected cell proportions can vary across tissues. Initial diagnostic testing typically uses DNA from blood leukocytes. However, using other tissues like buccal swabs, skin fibroblasts, or mesenchymal-origin cells from surgical samples can improve mosaicism detection. A negative result does not rule out BWS due to potential low-level mosaicism, rare balanced chromosomal rearrangements, or other unknown causes. In up to 20% of clinically diagnosed BWS cases, a molecular diagnosis remains elusive, possibly due to tissue mosaicism. Testing multiple tissues enhances diagnostic yield. Patients without molecular confirmation but with a clinical score ≥4 may still be diagnosed with classical BWS. For those with scores of 1–3, careful evaluation for alternative diagnoses is necessary.
Figure 2 outlines a diagnostic flowchart for BWS molecular diagnosis. First-line testing should assess IC1 and IC2 methylation levels and differentially methylated region (DMR) copy number. Methylation abnormalities are found in IC1 GOM, IC2 LOM, pUPD11 (showing both IC1 GOM and IC2 LOM), and CNVs. Methylation-specific multiplex ligation-dependent probe amplification (MS-MLPA) is a common first-line test, simultaneously detecting methylation percentage and DMR copy number. However, techniques with higher sensitivity for low-level mosaicism may be required, and testing multiple tissues is advisable in suspected mosaicism cases. To confirm pUPD, chromosomal microarray analysis (CMA), particularly SNP-based arrays, can detect low-level mosaicism and determine the pUPD region’s extent, which can inform management. Genome-wide pUPD (GWpUPD) may occur in up to 10% of pUPD 11p15 BWS cases, associated with additional clinical features and increased tumor risk. CDKN1C mutations are identified by gene sequencing, enabling cascade testing in families to determine recurrence risks.
Figure 2. Diagnostic Algorithm for Beckwith-Wiedemann Syndrome Molecular Diagnosis
Image alt text: Flowchart detailing the steps in molecular diagnosis of Beckwith-Wiedemann syndrome, starting from clinical scoring to various genetic tests and considerations for differential diagnosis and tissue mosaicism.
If CNVs are detected, chromosome microarray is recommended to characterize the deletion or duplication size and nature. Karyotyping or fluorescence in situ hybridization (FISH) can identify chromosomal translocations. In IC1 GOM patients, up to 20% may have small DMR CNVs, associated with higher recurrence risk, requiring targeted IC1 sequencing, especially in familial BWS cases. Deletions in IC2 are rare. About one-third of IC2 LOM patients may have multilocus imprinting disturbance (MLID), but its clinical significance is uncertain, making testing generally not indicated.
For BWS patients from multiple pregnancies, determining zygosity and chorionicity is crucial for diagnosis. For dizygotic dichorionic twins, no evaluation is needed for the unaffected twin. For monozygotic twins (both monochorionic and dichorionic), the unaffected twin should undergo clinical examination by a geneticist. Buccal swabs are preferred for discordant monozygotic twins to avoid aberrant methylation results in unaffected twins due to shared fetal circulation, which can occur in blood or saliva DNA.
Diagnostic Tests for Methylation Aberrations
Methylation-specific multiplex ligation-dependent probe amplification (MS-MLPA) is widely used as it detects both DMR methylation and copy number variations. However, it does not determine CNV size or content, making CMA or FISH more suitable for CNV characterization. MS-MLPA is PCR-based, using multiple probes in the 11p15 region and other genomic loci. Some 11p15 probes are methylation-specific, containing the Hha I restriction enzyme site in a CpG island. DNA samples are split into two aliquots: one for traditional MLPA to detect CNVs, and another for methylation-specific (MS) analysis. The MS aliquot includes Hha I, which degrades unmethylated sequences, amplifying only methylated DNA. Relative DNA amounts are quantified using fluorescently labeled primers to calculate the methylation index. MS-MLPA can also identify the parental origin of small CNVs, but CMA or FISH is recommended for further characterization. Controls from the same tissue type are crucial for accurate analysis. MS-MLPA offers high specificity and reliability for CNV and methylation status detection in the 11p15 region, with efficiency, low cost, and small DNA quantity requirements, making it ideal as a first-line diagnostic test.
For more sensitive detection of low-level mosaicism, quantitative methylation-sensitive PCR is used. This method involves sodium bisulfite treatment of genomic DNA followed by quantitative TaqMan PCR. Bisulfite converts unmethylated cytosines to uracil, while methylated cytosines remain unchanged. TaqMan probes with different fluorophores differentiate between methylated and unmethylated DNA, and the methylation index is calculated based on fluorescence intensity. Allele-specific methylated multiplex real-time quantitative PCR (ASMM RTQ-PCR) has shown higher sensitivity than MS-MLPA, detecting smaller methylation changes. Methylation-specific high-resolution melting (MS-HRM) detects methylation by analyzing melting profile differences between methylated and unmethylated DNA. After bisulfite treatment and PCR, a fluorescent dye is added, and fluorescence changes are monitored as DNA melts. Unmethylated alleles melt at lower temperatures than methylated alleles. MS-HRM has similar results and limitations to MS-MLPA and other methylation-specific PCR techniques.
Diagnostic Tests for Uniparental Disomy and Chromosomal Anomalies
Chromosomal microarray analysis (CMA), FISH, and karyotyping detect copy number variations, chromosomal abnormalities, and confirm uniparental isodisomy. CMA is the most common microarray technology for deletions and duplications but provides limited structural rearrangement information. FISH or karyotyping is often used with CMA for confirmation and translocation/insertion identification.
Two main types of CMA are comparative genomic hybridization (CGH) and single nucleotide polymorphism (SNP) analysis. Both detect submicroscopic changes undetectable by karyotyping by comparing hybridization intensities between patient and reference DNA. Labeled patient and reference DNA compete to hybridize to normal metaphase DNA. Equal hybridization indicates a normal sample, while imbalances suggest chromosomal aberrations. CGH detects deletions or duplications of a few kilobases, whereas SNP probes identify single nucleotide variations. CGH cannot detect balanced rearrangements, UPD, or low-level mosaicism.
SNP microarrays detect CNVs, copy-neutral loss of heterozygosity, and parental origin of CNVs to identify uniparental disomy. While MS-MLPA and methylation-sensitive PCR indirectly suggest pUPD when both IC1 and IC2 show abnormal methylation, SNP microarrays quantitatively determine UPD size based on affected SNPs. SNP microarrays are highly sensitive for UPD and mosaicism, distinguishing low-level mosaicism (1–5%) from normal samples using B-allele frequencies (BAF). They also more precisely differentiate UPD from chromosomal abnormalities than karyotyping or FISH. Genome-wide SNP arrays can distinguish mosaicism from chimerism (two cell lines with complete DNA sets). Microsatellite arrays, analyzing short tandem repeats (STRs), are similar to SNP microarrays in UPD detection but less sensitive.
Karyotyping identifies larger chromosome differences, detecting structural changes >3–10 Mb and complex rearrangements. Fluorescence in situ hybridization (FISH) detects gene structural changes with higher resolution than karyotyping. FISH uses fluorescent probes to bind specific DNA sequences, detecting their presence or absence. FISH requires prior knowledge of the abnormal region and allows only a few probes per test. While karyotyping and FISH differentiate chromosomal abnormalities from mosaic UPD, they cannot determine disomy or CNV size. After identifying chromosomal abnormalities, family testing is recommended as appropriate.
Diagnostic Tests for CDKN1C Mutations
Genetic sequencing is used to detect CDKN1C mutations and other gene mutations in the 11p15 region. This involves PCR amplification of the region of interest followed by Sanger sequencing to identify candidate mutations.
Prenatal Testing for BWS
Prenatal molecular diagnosis of BWS is possible in some cases, but negative results do not exclude the diagnosis due to mosaicism, necessitating postnatal confirmation. Genetic counseling should cover test benefits and limitations. Prenatal testing is indicated by abnormal ultrasounds showing omphalocele, macroglossia, or enlarged organs. Placental mesenchymal dysplasia, polyhydramnios, or elevated second-trimester alpha-fetoprotein (AFP) can also indicate BWS risk. Positive family history, particularly of CDKN1C mutations or chromosomal abnormalities, is another indication. Both native and cultured amniocytes can be used, though cultured cells may not accurately reflect fetal or placental genetic status. Methylation testing and CDKN1C sequencing are recommended for prenatal diagnosis. Chorionic villus sampling (CVS) may be limited by confined placental mosaicism, potentially not reflecting fetal (epi)genetic status. Negative or low-level changes in CVS require amniocentesis and/or postnatal testing. Maternal contamination is a risk, so parallel microsatellite analysis of maternal and fetal STRs is strongly advised. While a positive methylation or chromosomal test confirms BWS, a negative result does not exclude it due to complexity, mosaicism, and other undetected molecular alterations. Postnatal testing is essential for confirmation.
Management and Care Based on BWS Diagnosis
Determining the molecular subtype of BWS is crucial as genotype-phenotype correlations exist, influencing clinical presentation and tumor risk. Figure 3 shows facial photographs of patients with different BWS molecular subtypes. IC1 GOM patients tend to have high birth weight, organomegaly, and a high tumor incidence (28%), especially Wilms tumors. IC2 LOM patients typically present with omphalocele, macroglossia, nevus simplex, and have the lowest tumor risk (2.6%). pUPD patients often have lateralized overgrowth, hyperinsulinism, and intermediate tumor risk (16%). The correlation between phenotype severity and mosaicism or chromosomal isodisomy level is unclear. CDKN1C mutation patients share features with IC2 LOM, such as omphalocele and nevus simplex, with an estimated tumor risk of 6.9%, though data is limited.
Figure 3. Phenotypic Variability in Beckwith-Wiedemann Syndrome Subtypes
Image alt text: Facial photographs illustrating the phenotypic differences among patients with Beckwith-Wiedemann syndrome due to various molecular subtypes: IC2 LOM, IC1 GOM, chromosomal rearrangements, pUPD, GWpUPD, and CDKN1C mutation.
Prenatal Care
Prenatally diagnosed BWS requires management of specific congenital anomalies following standard protocols. BWS increases risks of polyhydramnios, gestational hypertension, pre-eclampsia, and preterm birth. Delivery and neonatal care should be planned accordingly. Post-delivery complications may include neonatal hypoglycemia, respiratory obstruction from macroglossia, and omphalocele surgical repair.
Hypoglycemia and Hyperinsulinism Management
Hypoglycemia affects 30–60% of BWS patients, caused by hyperinsulinism. It is usually transient, resolving within days, but persistent hyperinsulinism (HI) can occur. Neonates with suspected BWSp should be screened for hypoglycemia before discharge. HI may require diazoxide, somatostatin analogs (octreotide, lanreotide), or in severe cases, subtotal pancreatectomy. Congenital HI is more common in pUPD patients and requires endocrinologist management. HI in BWS can occur with or without mutations in beta-cell potassium channel genes (ABCC8 and KCNJ11) located near the BWS region on chromosome 11p.
Macroglossia Management
Macroglossia is present in about 90% of BWS patients, with approximately 40% undergoing tongue reduction surgery. Surgery need and timing depend on clinical status. Indications include respiratory issues, obstructive sleep apnea, feeding difficulties, drooling, speech and orthodontic problems. Airway evaluation and polysomnography assess obstructive sleep apnea. Early surgery may be needed for respiratory problems. Surgery before age 2–3 often yields good outcomes, including cosmetic improvement, tongue mobility, and no taste impairment.
Abdominal Wall Defects Management
Omphalocele and other abdominal wall defects in BWS are managed following standard protocols and local practices. No specific BWS-related recommendations exist.
Growth and Lateralized Overgrowth Management
Overgrowth occurs in 43–65% of BWS patients, and lateralized overgrowth is frequent in pUPD. Postnatal growth is usually in upper percentiles, slowing in late childhood. Lateralized overgrowth management depends on affected limbs. Leg length discrepancy (LLD) may require shoe lifts or surgical correction.
Tumor Screening Protocols for Early Detection
BWS is a cancer predisposition syndrome with about 8% overall tumor risk, varying by molecular subtype. Common tumor types include Wilms tumor (52%), hepatoblastoma (14%), neuroblastoma (10%), rhabdomyosarcoma (5%), and adrenal carcinoma (3%). Cancer risk is highest in the first 2 years, declining thereafter; no increased adult tumor risk is evident. IC1 GOM has the highest tumor risk (28%), followed by pUPD (16%), CDKN1C mutation (6.9%), and IC2 LOM (2.6%). IC1 GOM is primarily associated with Wilms tumor (95% of tumors in this group). IC2 LOM is more linked to hepatoblastoma, and CDKN1C mutations to neuroblastoma. pUPD patients have similar frequencies of Wilms tumor and hepatoblastoma; GWpUPD patients have similar tumor types as segmental pUPD but with increased hepatic/adrenal tumors into young adulthood. Cancer risk in BWSp clinical diagnoses or negative molecular testing requires further research.
Tumor screening protocols are recommended for early detection. American Association for Cancer Research Childhood Cancer Predisposition Workshop (AACR-CCPW) guidelines include abdominal ultrasound (USS) every 3 months until age 4, renal ultrasound every 3 months from age 4–7, and AFP screening every 3 months until age 4 for all BWSp patients. CDKN1C mutation patients should also have neuroblastoma screening (urine VMA/HVA and chest X-ray every 3 months until age 6, then every 6 months from age 6–10), plus abdominal imaging and AFP screening. An international consensus group recommends abdominal ultrasounds every 3 months until age 7 for high-risk BWSp subgroups: IC1 GOM, pUPD, CDKN1C mutation, other genomic rearrangements, and clinical BWS.
Serum alpha-fetoprotein (AFP) measurements could detect hepatoblastomas earlier than ultrasounds. However, AFP interpretation in infancy is complex due to variable levels and wide normal ranges. The consensus group does not routinely recommend AFP due to low hepatoblastoma incidence in BWSp and interpretation challenges.
AACR-CCPW recommends abdominal USS and AFP screening for all BWSp subtypes, using a 1% tumor risk threshold. While IC2 LOM has lower overall tumor risk, hepatoblastoma risk is increased, which has lower survival rates than Wilms tumor. Localized hepatoblastoma has high survival rates (80–100%), but late-stage tumors have poorer prognoses. AFP levels for hepatoblastoma screening should be clinically interpreted, as BWS patients have higher AFP levels in early childhood. AFP levels should decline over time and can be tracked with normograms. Significant AFP rises require repeat testing and imaging. AFP screening can differentiate hepatoblastoma from benign infantile hepatic hemangioma. Serial AFP monitoring can enable early hepatoblastoma detection, improving outcomes. Dried blood spot AFP measurement is a less invasive, accurate alternative to venipuncture.
Children with BWS and Wilms tumor have higher recurrence incidence and potential co-occurrence of progressive non-malignant renal diseases and bilateral Wilms tumor. Multifocal nephrogenic rests (nephroblastomatosis) can be difficult to distinguish from Wilms tumor and may require MRI. Partial nephrectomy and nephron-sparing strategies are advised for Wilms tumors. Nephro-urological anomalies occur in 28–61% of BWSp patients, including cortical and medullary cysts in about 10%, and increased hypercalciuria and nephrolithiasis. Adults with BWS should have a detailed clinical review and renal ultrasound at age 16 to guide ongoing surveillance. No clear association exists between BWSp and common adult-onset carcinomas, but more adult BWS research is needed.
Cardiac Features Management
Cardiac defects occur in 13–20% of BWS patients, with higher congenital heart disease incidence. Minor defects should be monitored by echocardiogram until resolution; severe defects may need surgery. IC2 CNVs and genomic rearrangements may predispose to long QT syndrome, requiring adult follow-up.
Cognitive and Neurological Features
Cognitive development is usually normal in BWS. Developmental delay may be associated with prematurity, severe hypoglycemia, unbalanced chromosomal rearrangements, and GWpUPD. Broader differential diagnoses should be considered in overgrowth disorders with learning disabilities without 11p15 anomalies.
Psychological Well-Being
BWSp diagnosis impacts patients and families psychologically and socially. Parents may be unprepared due to lack of family history. Tumor risk and surveillance can cause anxiety, though screening is generally perceived as helpful, not burdensome. Macroglossia can cause parental concern about peer interactions and emotional difficulties. Healthcare professionals should address psychosocial issues, referring families to genetic counselors, social workers, psychologists, or support groups.
Discussion
BWSp is a complex multisystem disorder from diverse molecular changes in the 11p15 region. Various genetic tests detect aberrant methylation and chromosomal abnormalities, guiding BWSp molecular diagnosis. Tissue mosaicism remains a diagnostic challenge, and negative tests do not exclude BWSp. A clinical BWS diagnosis is still valid with a score ≥4 even with negative molecular testing. However, molecular diagnosis is crucial for care coordination and management, and testing multiple tissues can improve diagnostic rates.
Author Contributions
KW: literature search, manuscript drafting, figure creation. JK: literature search, testing techniques section drafting, figure creation. KD: project conceptualization, manuscript editing. JMK: conceptualization, organization, manuscript editing.
Conflict of Interest
The authors declare no conflicts of interest.
Acknowledgments
The authors thank patients and families involved in BWS research and funding sources including Alex’s Lemonade Stand Foundation for Childhood Cancer; St. Baldrick’s Foundation Scholar Award; National Institutes of Health, Grant/Award Number: K08CA193915.
Footnotes
Funding. Alex’s Lemonade Stand Foundation for Childhood Cancer; St. Baldrick’s Foundation Scholar Award; National Institutes of Health, Grant/Award Number: K08CA193915.
References
[References] (The original article’s references should be listed here in markdown format)