1. Introduction
Cushing’s syndrome (CS) arises from excessive cortisol production. In most instances (80%), this is due to the over-secretion of adrenocorticotropic hormone (ACTH), often from a pituitary adenoma or, less commonly, a neuroendocrine tumor. Primary adrenal overproduction of cortisol accounts for the remaining 20%, with benign cortisol-secreting adenomas being the most frequent cause. Other adrenal CS etiologies, including adrenocortical carcinoma and bilateral adrenal hyperplasia, are less frequent, representing under 10% of adrenal CS cases [1]. Bilateral adrenal hyperplasia can occur in isolation or as part of a broader syndrome (Table 1). Morphologically, bilateral adrenal hyperplasia is categorized into primary bilateral macronodular adrenal hyperplasia (PBMAH) and micronodular forms, including primary pigmented micronodular adrenal dysplasia (PPNAD) and isolated micronodular adrenal hyperplasia (iMAD). Accurate Bilateral Adrenal Hyperplasia Diagnosis is crucial for effective patient management.
Table 1.
Germline defects linked to adrenal hyperplasia. Note: NA indicates Not Applicable; mutations described may only cause adrenal hyperplasia and have only been reported in case studies.
| Gene | Genetic | Function | Phenotype | Frequency of Adrenal Hyperplasia in Mutations |
|---|---|---|---|---|
| PRKAR1A | Unique inactivating mutations spread along the gene. 3 hotspots (c.709(−7–2)del6, c.491–492delTG, c82C > T).Large deletions described | Regulatory subunit R1α of PKA. PKA pathway inhibition | Isolated PPNAD (~12%)Carney complex: cardiac, skin and breast myxomas, lentigines, pituitary adenoma or hyperplasia (GH +/− PRL), LCCST, osteochondromyxoma, schwannomas | 26% to 60% [1,2,3] |
| PRKACA | Amplification of the gene | Catalytic subunit Cα of PKA. PKA pathway activation | PBMAHMacroglossia? | NA 1 |
| GNAS | Post-zygotic activating mutationsTwo hotspots (p.R201H and p.C174Y) | G protein subunit alpha stimulating. PKA pathway activation | Macronodular adrenal hyperplasiaMc Cune Albright syndrome: precocious puberty, Café-au-lait spot, polyostotic fibrous dysplasia, somatotroph adenoma or prolactinoma, multinodular goiter, hyperthyroidism | Near 5% [4,5] |
| PED8B PDE11A | Unique inactivating mutations | Phosphodiesterase type 8B and 11A. PKA pathway inactivation | iMAD | NA 1 |
| MC2R | Unique activating mutations | ACTH receptor. PKA pathway activation. | PBMAH | NA 1 |
| ARMC5 | Unique inactivating mutations spread along the gene. | Potentially control apoptosis and cell cycle.Interaction with PKA pathway and steroidogenesis? | PBMAHMeningioma (several cases described) | ND,High penetrance described in families |
| MEN1 | Unique inactivating mutations spread along the gene.Large deletions | Scaffold protein controlling gene transcription and many other cellular functions, such as proliferation | PBMAHPituitary adenomaPrimary hyperparathyroidismNeuroendocrine tumors | Case reports |
| FH | Unique inactivating mutations spread along the gene. | Krebs cycle | HLRCC: leiomyomatosis, renal cell cancer | Estimated at 0.8% [6] |
| APC | Unique inactivating mutations spread along the gene. | Wnt/β-catenin pathway inhibition | PBMAHFamilial adenomatous polyposis | Case reports |
The familial and bilateral nature of these conditions suggested a genetic basis, which is now confirmed in approximately 70% of PPNAD and 25% of PBMAH cases. Many genes involved in bilateral adrenal hyperplasia are tumor-suppressor genes. Knudson’s two-hit hypothesis explains that one allele is inactivated by a germline mutation (detectable in leukocytes), and the second allele is somatically inactivated (tumor-specific). Crucially, most genetic and molecular changes in these conditions activate the protein kinase A (PKA) pathway. The cAMP pathway, a fundamental intracellular signaling route, regulates cellular processes like proliferation, differentiation, and hormonal activity in endocrine tissues. In adrenal glands, ACTH binds to its MC2R receptor, activating the Gs subunit, which stimulates adenyl cyclase to produce cAMP. cAMP then activates PKA, which in turn phosphorylates targets like CREB, a transcription factor involved in adrenal proliferation and steroidogenesis. Phosphodiesterases degrade cAMP [2] (Figure 1A). Mutations in components of this pathway, or abnormal expression of G-protein coupled receptors, are frequent alterations in bilateral adrenal diseases (Figure 1B,C). The 2013 discovery of ARMC5 (armadillo repeat containing 5) alterations in PBMAH provided new insights into bilateral adrenal hyperplasia pathogenesis (Figure 1C).
Figure 1.
Alterations in the protein kinase A (PKA) pathway and ARMC5 in bilateral adrenal hyperplasia. (A) Normal adrenocortical cell function: ACTH activates MC2R, leading to Gα subunit activation. This activates adenylate cyclase (AC), converting ATP to cAMP. Phosphodiesterases (PDE) inactivate cAMP to AMP. PKA regulatory (R) subunits bind cAMP, releasing catalytic (C) subunits. These phosphorylate targets, including cAMP Response Element-Binding protein (CREB), activating steroidogenesis genes. ARMC5 blocks the cell cycle in G1 phase and induces apoptosis, degraded by Culin3. (B) PPNAD and iMAD: PKA pathway activation via (1) PRKAR1A mutations, (2) phosphodiesterase gene mutations, and (3) PRKACA catalytic subunit duplication. (C) PBMAH: PKA pathway activation by (1) local ACTH production, (2) MC2R mutations, (3) GNAS mutations, (4) aberrant G-protein coupled receptor expression, (5) phosphodiesterase gene mutations, (6) PRKACA catalytic subunit duplication, and (7) ARMC5 mutations leading to cell cycle activation and apoptosis loss. ARMC5 mutations can also prevent Culin3 binding and degradation. ARMC5 normally decreases PKA activity.
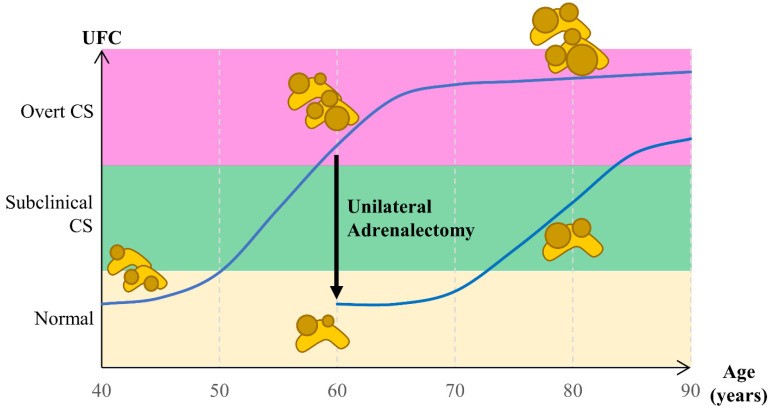
Management of bilateral adrenal hyperplasia lacks consensus due to its rarity, potential for subclinical CS, and risks of bilateral adrenalectomy (definitive adrenal insufficiency). Bilateral adrenalectomy was once standard for CS treatment, but unilateral adrenalectomy is now considered in PBMAH and PPNAD, showing promising outcomes, especially in PBMAH (Figure 2). This review summarizes current understanding of bilateral adrenal hyperplasia pathogenesis and treatment, emphasizing bilateral adrenal hyperplasia diagnosis.
Figure 2.
 Figure 2
Figure 2Cortisol secretion evolution in bilateral adrenal hyperplasia over time and the effect of unilateral adrenalectomy on cortisol levels.
2. Clinical Features and Diagnostic Approaches
2.1. Micronodular Adrenal Hyperplasia: Diagnostic Insights
Bilateral micronodular adrenal hyperplasia is defined by the presence of micronodules (3,4,5]. PPNAD is the most frequent endocrine manifestation of Carney complex (CNC), but in approximately 12% of patients, PPNAD occurs in isolation [6]. In contrast, isolated micronodular hyperplasia (iMAD) is characterized by hyperplasia of the internodular tissue [7], and its etiology is largely unknown. Diagnosis of micronodular adrenal hyperplasia relies on a combination of clinical evaluation, biochemical testing, and imaging.
PPNAD typically develops during the second and third decades of life, with a minority of cases presenting in the first three years [8]. Post-puberty, about 70% of PPNAD patients are female [6], although the reasons for this female predominance are unclear, estrogen’s role has been suggested.
Clinical presentation varies widely, even within families, ranging from subclinical to overt CS, with either sudden or gradual onset. Cyclic forms, characterized by rapid hypercortisolism followed by spontaneous remission, have also been reported [9,10]. Approximately 25% of CNC patients develop overt CS [8]. A key diagnostic indicator is the lack of cortisol suppression after a low-dose dexamethasone test, observed in 60% of CNC patients [6]. Interestingly, adrenal dysplasia has been found in autopsies of all CNC patients, even those without clinical symptoms [8], highlighting the importance of early bilateral adrenal hyperplasia diagnosis, even in subclinical cases.
Biochemical diagnosis of bilateral adrenal hyperplasia often reveals fluctuating urinary free cortisol (UFC) levels. A paradoxical cortisol elevation after a high-dose dexamethasone suppression test has been described, but with low sensitivity (39%) [11].
High-resolution computed tomography (CT) scans, both pre- and post-contrast, are valuable for bilateral adrenal hyperplasia diagnosis, potentially revealing micronodules as hypodense spots throughout the adrenal gland [12]. Macronodules can occasionally be present, particularly in older individuals [13].
Rarely, adrenocortical cancer [14,15], benign androgen-secreting adenoma [16], and pheochromocytoma [11] have been reported in PPNAD patients, but the causal relationship with PPNAD is unclear.
2.1.2. Carney Complex: Diagnostic Considerations
Carney Complex (CNC), first described in 1985 by J. Aidan Carney [3], is a rare, multiple neoplasia syndrome with diverse endocrine and non-endocrine manifestations [17]. Diagnosis of Carney Complex, and consequently PPNAD within CNC, relies on recognizing a constellation of clinical features. Prevalence is difficult to determine due to its rarity, but extensive series have studied over 350 cases [6]. Women represent approximately 60% of known patients [6,8], and all ethnic groups can be affected [8]. About 70% of cases are familial with autosomal dominant inheritance [6,8,19], the rest are considered sporadic. Mean age at diagnosis is 20 years [8], with rare diagnoses at birth (2%) [8]. Phenotypic variability within families is common [6,11].
Diagnostic criteria for CNC were established in 2001 [8]. Manifestations and their frequencies are listed in Table 2. Cardiac myxomas are a significant cause of morbidity and mortality in CNC. Myxomas can also occur in breasts and skin. Lentigines and blue naevi are frequent but less specific than cutaneous myxomas [10]. Pituitary abnormalities primarily involve the somatotroph and/or lactotroph axis, with acromegaly signs being uncommon and pituitary MRI often normal [20]. Thyroid manifestations include bilateral macronodules and papillary or follicular carcinomas. Large Cell Calcifying Sertoli Tumors (LCCST) are the most common gonadal lesions in males, while ovarian cysts are frequent in females [10,11]. Breast lesions, including adenomas and potentially carcinomas, are also observed [11]. Schwannomas in CNC are characterized by psammoma bodies and melanin, with malignancy reported in 10% of cases. Rare tumors include osteochondromyxomas and pancreatic tumors [10]. Diagnosis of PPNAD in the context of CNC requires considering these multi-systemic features.
Table 2.
Manifestations of Carney Complex.
| Clinical Features | Frequency (%) [1,2,3] | Age at Diagnosis (Years) [2,3] |
|---|---|---|
| PPNAD | 45–68 | Median: 25Bimodal age distribution: in the first 3 years of life or in the 2nd and 3rd decades |
| Skin lesion | ||
| Lentigines | 56–70 | From birth or appear progressively, fade after the 4th decade |
| Blue naevi | 17–50 | May appear in early childhood years |
| Cutaneous myxoma | 20–45 | May appear within the first 10 years of life |
| Cardiac myxoma | 23–53 | Median: 29Described in the first years of life |
| Hypersomatotropism | 10–19 | Median: 35 |
| Thyroid tumors | 5–25 | May appear within the first 10 years of life |
| Psammomatous melanotic schwannoma | 8–10 | Median: 35 |
| Osteochondromyxoma | 2–6 | Described in the first years of life but also in adults |
| Breast lesions | 19–42 | Breast myxomas may appear in childhood |
| LCCSCT | 33–41 | Median: 28Described from the first years of life |
2.2. Macronodular Adrenal Hyperplasia: Diagnostic Strategies
Primary bilateral macronodular adrenal hyperplasia (PBMAH) is characterized by multiple nodules larger than 1 cm in both adrenal glands. First described in 1964 [21], it affects both sexes, although sporadic cases may be more prevalent in females. PBMAH progresses slowly, with CS developing insidiously over years, leading to diagnosis typically after the fourth decade. Familial cases have been documented. Effective PBMAH diagnosis is essential for appropriate management.
PBMAH is considered rare, but its prevalence is likely underestimated. Many PBMAH cases are incidentally discovered during imaging for unrelated reasons. Adrenal incidentalomas occur in 1–5% of abdominal imaging, with 2.7–10% being bilateral. Subclinical CS is present in 35–40% of these bilateral incidentalomas, and a portion of these may represent PBMAH [22]. While some PBMAH cases are diagnosed due to overt CS, subclinical CS is more common. In larger series, subclinical CS may affect at least 50% of PBMAH cases [23]. The disease exhibits heterogeneity in cortisol over-secretion and morphology, even within families, posing challenges for bilateral adrenal hyperplasia diagnosis. Whether macronodular adrenal hyperplasia on imaging without biological abnormalities truly constitutes PBMAH remains an open question.
Differential diagnosis of bilateral adrenal hyperplasia must exclude conditions causing chronic ACTH overstimulation, such as Cushing’s disease, ectopic ACTH secretion, and congenital adrenal hyperplasia [24]. NR3C1 gene mutations, encoding the glucocorticoid receptor, have been identified in 5% of bilateral adrenal incidentaloma cases. These patients exhibit cortisol resistance, elevated UFC, and unsuppressed cortisol post-dexamethasone, but lack CS features. They may develop hypertension due to altered 11β-hydroxysteroid dehydrogenase type 2 activity [25].
Current guidelines for PBMAH imaging follow-up are lacking. The European Endocrine Society recommends individual follow-up for adrenal incidentalomas larger than 4 cm or with spontaneous density above 10 HU (common in PBMAH), with repeat imaging at 6 months [26]. Given the slow progression of PBMAH, hyperplasia stability is likely. Adrenal carcinoma has not been reported in PBMAH. Further imaging is guided by hypercortisolism progression, clinical impact, and treatment decisions.
2.2.2. Multiple Tumor Syndromes Associated with Macronodular Adrenal Hyperplasia: Diagnostic Context
MEN1: Multiple endocrine neoplasia type 1 (MEN1), an autosomal dominant disorder linked to MEN1 gene mutations (11q13), includes primary hyperparathyroidism (95%), pancreatic neuroendocrine tumors (50%), pituitary adenomas (40%), and thymic carcinoid tumors [27]. Adrenal lesions (hyperplasia or nodules) are reported in up to 50% of MEN1 patients [28,29,30], but Cushing’s syndrome of adrenal origin is rare (0.6% in a French cohort [28]). PBMAH has been described in two MEN1 patients [28,31]. Mouse models support the link between MEN1 and adrenal tumors [32]. Diagnosis of PBMAH in MEN1 requires considering the broader clinical picture.
Fumarate Hydratase: Hereditary leiomyomatosis-kidney cancer syndrome (HLRCC), caused by autosomal dominant fumarate hydratase (FH) gene mutations (1q43), involves the Krebs cycle enzyme FH. Ten HLRCC patients with PBMAH treated by adrenalectomy have been reported [33,34]. Loss of heterozygosity (LOH) in one PBMAH patient [33] and a germline FH mutation in a sporadic PBMAH case [35] support a causal link. Diagnosis of PBMAH in HLRCC is important for comprehensive patient care.
Familial polyposis coli: Familial polyposis coli or Gardner’s syndrome, due to APC gene mutations, features multiple colonic polyps and early-onset colon cancer. Other manifestations include pigmented retinal lesions, desmoid tumors, osteomas, thyroid nodules, and other malignancies [36]. PBMAH has also been described in these patients [35,37,38], with somatic events at the APC locus supporting causality [37,38]. Diagnosis of PBMAH in familial polyposis coli is relevant for risk assessment and management.
Beckwith–Wiedemann syndrome: Beckwith–Wiedemann syndrome, an imprinting disorder due to genetic or epigenetic alterations at 11p15.5 (including H19, IGF2, CDKN1C genes), includes overgrowth, neonatal hypoglycemia, congenital malformations, and embryonic tumor predisposition [39]. While adrenocortical carcinoma is the most frequent adrenal complication, CS due to bilateral nodular adrenal enlargement has been reported in infants. Histology shows adrenal cortex cytomegaly, a pathognomonic feature [40]. Diagnosis of PBMAH in Beckwith–Wiedemann syndrome is crucial in pediatric cases with CS.
3. Pathogenesis of Bilateral Adrenal Hyperplasia and Diagnostic Implications
3.1. Alterations in the PKA Pathway: Diagnostic Relevance
3.1.1. PRKAR1A Alterations in PPNAD: Genetic Diagnosis
Genetic studies in CNC patients have identified loci at chromosomes 17p22–24 and 2p16. No candidate gene has been found at chromosome 2 [41]. On chromosome 17, the PRKAR1A gene, encoding the R1α regulatory subunit of protein kinase A, was identified in 2000 as the causal gene [42,43]. Genetic diagnosis of PPNAD, and CNC, frequently involves PRKAR1A mutation analysis.
PRKAR1A is a tumor-suppressor gene with autosomal dominant transmission. Mutations are distributed across its 10 coding exons and splicing-essential intronic sequences, mainly single-base substitutions and short deletions [6,44]. Large deletions are less common [45].
In 80% of cases, mutations cause premature stop codons, leading to mutant mRNA degradation and haploinsufficiency [6,44,46]. Other mutations result in altered proteins with dominant negative effects, potentially negating the need for a second somatic event [46]. PRKAR1A mutations are found in 80% of CNC families and 37% of sporadic cases [6]. Hotspot mutations include c82C > T (exon 2), c.491_492delTG (exon 5), and c.709(−7–2)del6 (intron 7) [6]. Penetrance is approximately 95% by age 50, with incomplete penetrance for c.709(−7–2)del6 and c.1A > G mutations [44]. Genotype-phenotype correlations exist [6]. Genetic testing for PRKAR1A mutations is a key component of PPNAD diagnosis, especially in CNC context.
Mouse models of Prkar1a inactivation demonstrate thyroid tumors, schwannomas, bone lesions in heterozygotes [49] and adrenal hyperplasia, thyroid hyperplasia/adenomas, and other tumors with biallelic inactivation [50]. Adrenal-specific inactivation in female mice leads to CS, adrenal hyperplasia, and zonation defects [51], with androgens protecting males [52]. PKA pathway crosstalk with Wnt/β-catenin and mTOR pathways contributes to tumorigenesis [2]. Pigmentation in nodules is due to impaired autophagy and lipofuscin accumulation [53]. These findings reinforce the importance of PKA pathway dysregulation in bilateral adrenal hyperplasia diagnosis and pathogenesis.
3.1.2. Other PKA Pathway Gene Alterations: Diagnostic Expansion
PRKACA: Germline PRKACA amplification (duplication/triplication) has been found in 5 of 35 bilateral adrenal hyperplasia patients, including PPNAD and PBMAH cases [54,55]. This overexpression of catalytic subunits increases PKA activity [54]. Genetic diagnosis may include PRKACA amplification screening, particularly in PBMAH and PPNAD.
MC2R: MC2R mutations, affecting the ACTH receptor, have been reported in two sporadic PBMAH cases [56,57], but their definitive role is still under investigation.
Phosphodiesterases: PDE11A variants appear to predispose to PBMAH [58,59]. In vitro studies show increased cAMP levels and PKA activity with these variants [59]. Mutations in PDE11A and PDE8B genes are implicated in iMAD in children [60,61]. PDE11A variants may also increase susceptibility to LCCST and PPNAD in CNC patients, especially men [62]. Genetic diagnosis could consider PDE11A and PDE8B variants, especially in PBMAH and iMAD.
GNAS: McCune Albright syndrome (MAS), caused by post-zygotic GNAS mutations, is the most common cause of adrenal CS in children [63]. It involves fibrous dysplasia, café au lait spots, and endocrine activation, including CS due to bilateral adrenal hyperplasia, often presenting in the first year of life. Histology shows bi-morphic hyperplasia with cortical atrophy and fetal cortex persistence [64]. Somatic GNAS mutations are also found in PBMAH nodules [65]. Diagnosis of MAS-related bilateral adrenal hyperplasia in children relies on clinical features and genetic testing for GNAS mutations.
3.1.3. Aberrant G-Coupled Protein Receptor Expression in PBMAH: Functional Diagnosis
Abnormal cortisol secretion due to G-coupled protein receptors other than MC2R was an early pathogenic mechanism identified in PBMAH. Food-dependent CS, due to abnormal gastric inhibitory polypeptide (GIP) receptor expression, was described in 1992 [66,67]. These patients often exhibit fasting hypocortisolism [66,67]. Abnormal cortisol responses to various stimuli suggest aberrant expression of different receptors [68], including eutopic (normally expressed) and ectopic (abnormally expressed) receptors. Functional diagnosis of PBMAH can involve testing for these aberrant receptor responses.
Eutopic receptors include vasopressin V1, luteinizing hormone/human chorionic gonadotropin (LH/HCG), serotonin 5-HT4, and leptin receptors. Ectopic receptors include GIP, vasopressin V2 and V3, serotonin 5-HT7, glucagon, beta-adrenergic, and angiotensin II AT1 receptors. Diagnostic tests using specific ligands can assess the presence of these receptors (Table 3). In one series, 87% of PBMAH patients had at least one abnormal response, most commonly to posture (67%), metoclopramide (56%), and glucagon (47%) [70]. Food-response was less frequent (12%) [70]. GIP and LH/HCG receptor abnormalities can cause CS during pregnancy or post-menopause [71]. Abnormal responses in bilateral adrenal incidentaloma patients can support PBMAH diagnosis, although they can also be seen in other adrenal tumors [68,72].
Table 3.
Aberrant G-coupled protein receptor expression in PBMAH and diagnostic protocols. Adapted from [69,70,71]. A plasma cortisol change > 25% from baseline after stimulation defines a response (25-49%: partial, >50%: positive).
| Receptor | Ligand | Diagnostic Tests |
|---|---|---|
| Ectopic receptors | ||
| GIP receptor | GIP | Standard mixed meal, IV GIP infusion |
| V2R/V3 receptor | AVP/Anti-diuretic hormone | Supine-to-upright posture test, AVP/IM/SC desmopressin infusion (terlipressin) |
| β-adrenergic receptor | β-epinephrine | Insulin hypoglycemiaIV isoproterenol infusion |
| AT1 receptor | Angiotensin 2 | Supine-to-upright posture test,IV angiotensin 2 infusion |
| 5-HT7 receptor | Serotonin | Metoclopramide administration |
| Glucagon receptor | Glucagon | IV glucagon infusion |
| Eutopic receptors | ||
| V1R receptor | AVP/Anti-diuretic hormone | Supine-to-upright posture test IM desmopressin infusion (terlipressin) |
| 5-HT4 receptor | Serotonin | Metoclopramide administration |
| LH/HCG receptor | LH/HCG | IV GnRH infusionIM LH or HCG infusion |
| PRL receptor | Prolactin | Chlorpromazine administrationIV TRH infusion |
AVP: Arginine Vasopressin, AT1 receptor: Angiotensin 2 Type 1 receptor, GnRH: Gonadotropin-Releasing Hormone, PRL: Prolactin, TRH: Thyrotropin-Releasing Hormone.
Aberrant receptor expression/overexpression has been confirmed by qPCR [68] and transcriptomic analysis [73,74]. It typically activates the PKA pathway. Primary adrenocortical cells from patients with abnormal cortisol responses show increased cortisol production upon ligand stimulation [66,68,75,76,77,78]. Xenograft studies using bovine adrenocortical cells expressing GIP or LH/HCG receptors induced graft hyperplasia and CS in immunodeficient mice [79,80]. The mechanisms causing aberrant receptor expression are largely unknown [68], potentially linked to ARMC5 inactivation [82]. Somatic duplication of the GIP receptor locus has been seen in food-dependent cortisol-secreting adenomas and PBMAH [83]. Transcriptomic analysis suggests common molecular alterations in food-dependent Cushing PBMAH [84]. Functional testing for aberrant receptors is a valuable adjunct to PBMAH diagnosis.
3.2. ARMC5 Mutations in PBMAH: Genetic Diagnosis and Prognostic Implications
3.2.1. Genetic Mutations of ARMC5: Diagnostic and Familial Screening
In 2013, ARMC5 (Armadillo repeat containing 5) mutations were identified as responsible for PBMAH [85]. LOH at chromosome 16p was a frequent finding in adrenal tumor tissues. Whole-genome and Sanger sequencing identified ARMC5 at 16p as the PBMAH gene in a 33-patient series [85]. Subsequent studies confirmed ARMC5 mutations in approximately 25% of PBMAH cases, with potentially higher prevalence in Japan [23,86,87,88,89]. Genetic diagnosis of PBMAH now routinely includes ARMC5 mutation screening.
ARMC5 mutations correlate with more severe disease, higher hypercortisolism, larger adrenal hyperplasia, and more nodules [23]. Patients more frequently present with hypertension [23], and ARMC5 variants are linked to low renin hypertension, higher glucose, and HbA1c in African Americans [90,91]. Cortisol and aldosterone co-secretion has been reported [90]. ARMC5-mutated patients more often undergo surgery [23]. Food response is absent in ARMC5 patients, while vasopressin or orthostatism responses can be observed [23,92,93]. ARMC5 mutation status provides prognostic information and guides treatment decisions.
ARMC5 mutations account for approximately 80% of familial PBMAH [88,89,92,93,94]. Penetrance is high but incomplete [88,92], with variable phenotypes even in relatives, sometimes limited to subtle adrenal CT changes or pituitary-adrenal axis alterations [88,94]. Meningiomas have been reported in ARMC5-mutated patients [88,93,94,95,96], with LOH or second allele mutations in meningeal tumors supporting causality [94,95]. Familial screening for ARMC5 mutations is recommended in PBMAH cases.
ARMC5 inactivation follows the two-hit model for tumor-suppressor genes. Mutations are scattered along coding sequences, mostly unique to families, with no clear hotspots, although some mutations have been recurrently identified [97]. Gene deletions are less common [23,98].
3.2.2. Function of ARMC5: Mechanistic Insights Relevant to Diagnosis
The function of ARMC5 was unknown upon its identification as a PBMAH gene in 2013. ARMC5 is part of the Armadillo repeat protein family, containing armadillo repeat and BTB/POZ domains for protein-protein interaction. It is ubiquitously expressed [99]. Initial studies suggested ARMC5 involvement in apoptosis, with mutant ARMC5 overexpression in adrenocortical cell lines leading to apoptosis loss [23,85,100].
ARMC5 inactivation in vitro reduces steroidogenesis and cortisol production gene expression [85,100], consistent with transcriptome analysis showing reduced steroidogenic enzyme expression [101] and decreased cortisol production in PBMAH cell cultures [73]. CS may arise when adrenal mass compensates for reduced cellular steroidogenesis [97]. Adrenal gland size correlates with 17-hydroxycorticosteroids in ARMC5 variant carriers [102]. These functional insights are important for understanding bilateral adrenal hyperplasia diagnosis and progression.
Armc5 knockout mice are embryonically lethal [82,103]. Heterozygotes (Armc5+/−) develop hypocorticosteronemia at 12 months, supporting in vitro findings of reduced steroidogenesis. Prkaca expression is decreased in these mice [99]. Similarly, reduced PRKACA expression and PKA activity are seen in PBMAH nodules [104]. Hypocorticosteronemia in Armc5+/− mice is transient, with some developing hypercorticosteronemia at 18 months. Armc5+/− mice show cortex damage, but not macronodules [99], while adrenal hyperplasia is seen in Armc5−/− mice [103].
ARMC5 also regulates cell cycle. It interacts with Cullin 3 via its BTB/POZ domain, leading to ARMC5 degradation. ARMC5 overexpression alters G1-S progression, blocked by Cullin 3. BTB domain mutations affect ARMC5 degradation and cell cycle action [105]. ARMC5 involvement in T-cell function is also suggested [103].
3.3. Paracrine and Autocrine Factors in PBMAH: Potential Diagnostic Markers
Paracrine and autocrine regulation of adrenal glands by peptides or neurotransmitters from chromaffin cells, nerve endings, or immune cells is established [106,107,108]. Chromaffin cells produce ACTH locally [109]. In PBMAH, specific cortical cell clusters also produce ACTH, expressing proopiomelanocortin and proconvertase 1, with a steroidogenic, gonadal-like differentiation [110]. Adrenal venous catheterism confirmed adrenal ACTH production in PBMAH. ACTH immunostaining correlates with cortisol levels [110]. These ACTH-producing cells likely contribute to CS in PBMAH and explain why ACTH levels are often not suppressed despite hypercortisolism. This led to replacing “ACTH-independent macronodular adrenal hyperplasia” with PBMAH [111]. The origin of these cells and ARMC5 role are unclear. Further understanding of paracrine factors may reveal novel diagnostic markers for bilateral adrenal hyperplasia.
Local serotonin production by perivascular mast cells in the adrenal cortex subcapsular area stimulates aldosterone production [108,112]. In PBMAH, serotonin-producing cell clusters in nodules, along with aberrant 5-HT4 and 5-HT7 receptor expression, suggest an abnormal 5-HT paracrine pathway contributing to cortisol hypersecretion [75,108]. Chromogranin A [110] or AVP [75] expressing cells in PBMAH suggest local stimulation of β or α2-adrenergic and AVP receptors [108].
4. Treatment Strategies Following Bilateral Adrenal Hyperplasia Diagnosis
4.1. Treatment Decisions in Bilateral Adrenal Hyperplasia: Balancing Risks and Benefits
Treatment to normalize cortisol levels is generally indicated in overt CS [113]. However, bilateral adrenalectomy for bilateral adrenal hyperplasia induces definitive adrenal insufficiency, requiring lifelong hormone replacement and posing risks of adrenal crisis. Definitive adrenal insufficiency also increases morbidity and mortality from cardiovascular disease and infections [114,115,116]. As CS in bilateral adrenal hyperplasia is often subclinical, the benefit/risk balance is complex. The benefit of surgical treatment for subclinical CS adrenal incidentalomas versus medical management of cardiovascular risk factors remains unclear [117]. Meta-analysis suggests improved cardiovascular risk factors with adrenalectomy for subclinical CS [118], but prospective studies are needed.
Whether adrenal incidentaloma study results apply to bilateral adrenal hyperplasia is uncertain. PPNAD onset is typically younger than adrenal incidentaloma diagnosis, necessitating careful consideration of long-term consequences of adrenal insufficiency versus subclinical CS. Unilateral adrenalectomy emerges as a promising option for bilateral adrenal hyperplasia (Figure 2). Accurate diagnosis of bilateral adrenal hyperplasia is essential for guiding appropriate treatment decisions.
4.2. Surgical Treatment Options
4.2.1. Surgical Treatment of PPNAD: Unilateral vs. Bilateral Adrenalectomy
Bilateral adrenalectomy was historically the standard treatment for overt CS and adrenal hyperplasia [113]. In adult PPNAD patients, bilateral adrenalectomy is still often preferred for complete CS remission. Incomplete resection can lead to disease persistence [119].
Unilateral adrenalectomy has been proposed to avoid definitive adrenal insufficiency in PPNAD. A review of published cases showed initial success in 66% of patients (32/48) [120], mostly with overt CS. Side selection was based on macronodules or asymmetry on 131I-norcholesterol scintigraphy. Only 7 of 32 initially remitted patients required contralateral adrenalectomy for CS recurrence. Among the 25 without further surgery, follow-up was incomplete in some cases [121,122,123,124,125,126]. Only 12 patients (48% of initial remissions) remained recurrence-free after 16-113 months follow-up [127,128].
Unilateral adrenalectomy may be an option in selected PPNAD patients, especially younger ones. However, data interpretation requires caution due to variable remission criteria and potential publication bias towards successful cases. In a larger series, only 35% achieved initial remission. UFC levels decrease after unilateral adrenalectomy, reflecting reduced adrenal mass and reverting to levels seen 10-20 years prior.
4.2.2. Surgical Treatment of PBMAH: Unilateral Adrenalectomy as a Preferred Approach
Given the later onset and higher subclinical CS frequency in PBMAH compared to PPNAD, unilateral adrenalectomy is particularly appealing. Proposed since the late 1990s, 117 PBMAH patients across 23 case reports and series have been reported to undergo unilateral adrenalectomy [120]. Initial remission was achieved in 93% of patients, with remission duration varying from months to 15 years. Only 15% experienced recurrence, undergoing contralateral adrenalectomy after a median of 72 months [120]. Despite retrospective study limitations, unilateral adrenalectomy appears effective for most PBMAH patients.
UFC normalizes in nearly all patients, with an approximately 8-fold reduction one month post-surgery, while adrenal size reduces by roughly half. This discrepancy may be due to corticosteroid-binding globulin saturation at high cortisol levels, leading to a greater fractional reduction in free cortisol. Adrenal insufficiency due to corticotroph deficiency occurs in about one-third of patients, potentially persisting for years [120]. The Synacthen test may yield false positives post-unilateral adrenalectomy due to ACTH overactivation of remaining hyperplastic tissue [130].
Removal of the larger adrenal is typically performed, consistent with 131I-norcholesterol scintigraphy showing maximal uptake in larger glands [130]. Adrenal venous sampling is less frequently used due to invasiveness and specialist center requirements [131,132].
Improvements in cortisol-related comorbidities like obesity, diabetes, and hypertension are commonly reported post-unilateral adrenalectomy [130,133]. However, a recent study suggests potentially insufficient biochemical remission compared to bilateral adrenalectomy in some patients, with persistent elevations in post-dexamethasone cortisol or midnight salivary cortisol [133]. Adrenal-sparing surgery, removing one adrenal and a portion of the other, has been proposed, but its benefit over unilateral adrenalectomy needs evaluation [134]. Careful post-operative monitoring is crucial following any surgical treatment for bilateral adrenal hyperplasia.
4.3. Medical Treatment Options
All bilateral adrenal hyperplasia patients with subclinical CS require regular screening, comorbidity management, and cardiovascular risk factor control. Aberrant receptors offer potential for targeted medical therapy. Beta-blockers, particularly propranolol, have been used for posture-responsive cortisol secretion, with long-term control reported, but side effects are limiting [135,136,137]. Somatostatin analogs can be initially effective in food-dependent CS, but escape is common [135,138,139]. Leuprorelin, a GnRH agonist, has shown long-term response in LH/HCG-responsive PBMAH [139]. Multiple receptor responses in a patient can limit monotherapy efficacy.
Anti-cortisolic drugs can be used temporarily in overt CS patients awaiting surgery. Long-term steroidogenesis inhibitors like ketoconazole, metyrapone, or mitotane have been proposed for PBMAH [140,141] and PPNAD [142] with overt CS. Metyrapone at bedtime may restore circadian rhythm in subclinical CS adrenal incidentalomas [143]. Osilodrostat, a potent 11β-hydroxylase inhibitor, is a potential long-term alternative for PBMAH and PPNAD, but further studies are needed to confirm long-term efficacy and tolerability. Medical management plays a crucial role in bilateral adrenal hyperplasia, both as primary and adjunctive therapy.
5. Conclusions and Future Directions in Bilateral Adrenal Hyperplasia Diagnosis
Significant progress in understanding bilateral adrenal hyperplasia pathogenesis has occurred in recent decades. The PKA pathway is central, particularly in PPNAD, with alterations leading to CS and slow-growing hyperplasia. ARMC5 mutations are a key discovery in PBMAH pathogenesis. ARMC5 function, involving cell cycle, proteasomal degradation, and PKA pathway crosstalk, is complex and requires further investigation. Whole-genome sequencing may identify new genes in unresolved micronodular adrenal hyperplasia (30%) and PBMAH (75%) cases. Understanding paracrine regulation will also advance knowledge of these diseases.
Genetic screening should be offered to all bilateral adrenal hyperplasia patients, including PRKAR1A analysis in PPNAD and ARMC5 in PBMAH. Phosphodiesterase genes and PRKACA analysis should be considered. Next-generation sequencing facilitates comprehensive gene screening, including PRKACA amplification. Genetic counseling is essential for PRKAR1A mutation carriers and should be discussed for ARMC5 carriers.
Bilateral adrenalectomy is effective, but clinicians must weigh adrenal insufficiency risks against hypercortisolism complications, especially in subclinical CS. Unilateral adrenalectomy is a compelling alternative, particularly in PBMAH. Prospective studies are needed to compare medical therapy and adrenalectomy for subclinical CS, with both short and long-term evaluations of hypercortisolism complications. Continued research into bilateral adrenal hyperplasia diagnosis, pathogenesis, and treatment is essential for optimizing patient outcomes.
Acknowledgments
We acknowledge Omolara Khadija Tijani for the English editing.
Author Contributions
B.C.: writing; S.E.: original draft preparation, writing—review and editing; M.-C.V.: review and editing. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
Footnotes
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.