Eosinophilic disorders present a significant diagnostic conundrum in clinical practice due to their rarity and varied clinical manifestations. The classification of these disorders has historically been inconsistent, sometimes based on the location of eosinophil infiltration and at other times on peripheral blood eosinophil counts. This article delves into the complexities of diagnosing eosinophilic diseases through a detailed case report of a 54-year-old woman with a history of asthma who presented with bilateral lung infiltrates, shortness of breath, and eosinophilia. The differential diagnosis in such cases is extensive, encompassing infectious etiologies, inflammatory conditions like Churg–Strauss syndrome, and hematologic disorders such as hypereosinophilic syndrome. We aim to elucidate the inherent diagnostic challenges and discuss the evolving diagnostic landscape shaped by advancements in genetic diagnostics.
Case Presentation
A 54-year-old Caucasian woman with a history of asthma was admitted to our emergency department with a 3-week history of productive cough, dyspnea, and malaise. Her symptoms began four weeks prior with ear pain and nasal congestion, leading to a diagnosis of acute sinusitis by her family physician. She was initially treated with a 10-day course of levofloxacin and triamcinolone nasal spray. However, her condition worsened, with increasing exertional dyspnea followed by fever, chills, rigors, and intermittent night sweats.
Three days before presenting to our emergency department, she was re-evaluated by her family physician and diagnosed with community-acquired pneumonia. Clarithromycin (500 mg twice daily) was prescribed; however, her symptoms continued to progress, prompting her emergency department visit for further investigation.
The patient lived with her husband and adult child. Her occupation as director of a homeless shelter program involved weekly inspections of shelters. A tuberculin skin test conducted 8 years prior was negative. She denied HIV risk factors, was a lifelong non-smoker, and consumed 5 standard alcoholic beverages per week. She reported no sick contacts or animal allergies and had no recent travel history.
Her medical history was notable for allergic rhinitis, recurrent sinusitis, and asthma. Her asthma management included inhaled corticosteroids (fluticasone), long- and short-acting β2-agonists (salbutamol and salmeterol), and a leukotriene receptor antagonist (montelukast), which she had started 8 months before presentation. She had required systemic corticosteroids for asthma exacerbations twice in the preceding 6 months. Her current medications were her asthma regimen, clarithromycin, and triamcinolone nasal spray. She had a known anaphylactic allergy to lactose. There was no family history of respiratory or inflammatory diseases.
Upon examination, the patient appeared thin and exhibited moderate respiratory distress at rest. She was febrile (38°C), with a pulse rate of 104 beats/min, blood pressure of 139/77 mm Hg, respiratory rate of 34 breaths/min, and an oxygen saturation of 92% on room air. Chest auscultation revealed diminished breath sounds throughout both lung fields, with crackles predominantly in the upper lobes bilaterally. Cardiac examination showed normal heart sounds and a grade II/VI systolic ejection murmur at the heart base. No rash was present. The rest of the physical examination was unremarkable.
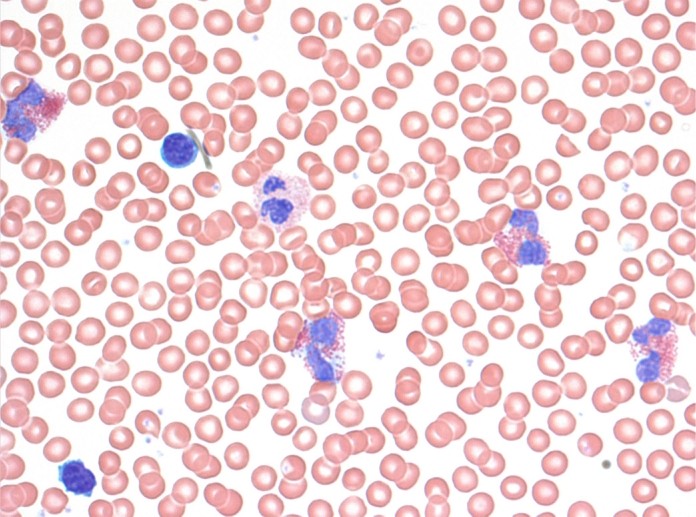
Initial blood work revealed a hemoglobin level of 146 g/L (normal range 138–175) and a significantly elevated white blood cell count of 29.3 × 109/L (normal 4.5–11), with 16.7 × 109/L (57%) eosinophils (normal 0%–3%), 37.8% neutrophils (normal 50%–62%), 3% lymphocytes (normal 25%–40%), 2% monocytes (normal 3%–7%), and no basophils (normal 0%–1%) (Fig. 1). Arterial blood gas analysis showed a pH of 7.49 (normal 7.35–7.45), PCO2 of 35 mm Hg (normal 35–45), PO2 of 57 mm Hg (normal 80–100), and bicarbonate level of 27 mmol/L (normal 22–26). Renal function was normal. Lactate dehydrogenase was elevated at 357 IU/L (normal 100–195), and C-reactive protein was markedly high at 136 mg/L (normal <10).
Radiographic findings from a chest X-ray (Fig. 2) were suggestive of pneumonia. Given her occupational history and the subacute nature of her illness, tuberculosis was considered. However, her asthma history and the presence of bilateral pulmonary infiltrates strongly suggested Churg–Strauss syndrome as a high probability in the differential diagnosis (Box 1).
Empirical broad-spectrum antibiotic treatment with ceftriaxone and clarithromycin was initiated, and montelukast was discontinued due to its potential association with Churg–Strauss vasculitis in asthmatic patients. Despite antibiotics, the patient remained febrile (39°C), tachycardic (110 beats/minute), and tachypneic (40 breaths/min), requiring 35% oxygen to maintain oxygen saturation above 90%. Blood, sputum, urine, and stool cultures were negative. Three sputum samples were negative for acid-fast bacilli, and a urine ELISA for Legionella antigen was also negative.
A contrast-enhanced computed tomography (CT) scan of the chest revealed mild mediastinal lymphadenopathy, with right paratracheal lymph nodes measuring 1.4 cm. Small bilateral pleural effusions and extensive airspace disease with scattered consolidation in all lobes, becoming confluent in the upper lobes, were noted. There was no evidence of pulmonary embolism, cavitation, loculation, or pleural irregularity (Fig. 3).
On the third hospital day, the patient reported worsening dyspnea and retrosternal chest heaviness at rest. Her troponin T level was elevated at 0.94 μg/L, creatine phosphokinase (CPK) was 290 IU/L (normal 5–160), and the CPK MB fraction was 9%. Electrocardiogram (ECG) showed anterior ST-segment elevation and mild PR-segment depression, suggesting ischemia or pericarditis. Echocardiography revealed normal left ventricular size, an ejection fraction of 50% (normal > 60%), anterior wall hypokinesis, trace pericardial effusion, and no valvular abnormalities. These findings, along with negative anti-neutrophil cytoplasmic antibodies (ANCA) and marked eosinophilia, pointed towards myocardial involvement secondary to Churg–Strauss syndrome or hypereosinophilic syndrome.
Bronchoscopy was performed, revealing no discrete lesions. Bronchial lavage fluid analysis showed 92% eosinophils (normal <1%). Bone marrow aspiration and biopsy demonstrated 45%–55% eosinophilic hyperplasia (normal 1.5%), with normal erythropoiesis and granulopoiesis. No nuclear atypia, granulomata, lymphoid aggregates, or metastatic cells were seen (Fig. 4).
To further investigate Churg–Strauss syndrome, a CT scan of the paranasal sinuses revealed minor mucosal thickening at the floor of the maxillary sinuses.
Considering the systemic eosinophilia, multisystem involvement with prominent pulmonary symptoms, history of asthma, and lack of response to antibiotics, the primary working diagnoses were Churg–Strauss syndrome and hypereosinophilic syndrome.
Antibiotics were discontinued, and prednisolone 50 mg once daily was initiated. Over the next 5 days, her cough, fever, and dyspnea resolved. White blood cell count and differential returned to normal. A repeat chest radiograph showed improvement in bilateral lung opacities, and a repeat ECG was negative for ischemia.
The patient was discharged with a slow prednisolone taper to 20 mg daily. The presumptive diagnosis at discharge was Churg–Strauss syndrome. Outpatient echocardiography confirmed resolution of wall motion abnormalities. Cytogenetic studies for hypereosinophilic syndrome were negative for the F1P1L1-PDGFR-alpha mutation, although it’s noted that the positivity rate varies widely (4%–60%). A positive result would predict imatinib treatment success, with remission rates of 60%–80%. Interleukin-5 levels, sometimes elevated in clonal hypereosinophilia, were also normal, suggesting anti-interleukin-5 therapy would likely be ineffective.
Three months post-discharge, recurrent respiratory symptoms and increased blood eosinophil count developed. Hydroxyurea was started, leading to normalization of eosinophil counts. Prednisolone was tapered to 10 mg/day. Cardiac MRI showed mild left ventricular dilation with normal ejection fraction and systolic function. Other cardiac structures were normal. Repeat chest radiography and abdominal ultrasound were unremarkable.
Differential Diagnosis of Bilateral Lung Infiltrates in Eosinophilic Disorders
The initial presentation of this patient mimicked community-acquired pneumonia, highlighting the broad differential diagnosis for bilateral lung infiltrates, particularly when coupled with eosinophilia. When evaluating bilateral lung infiltrates, especially in the context of eosinophilia, a systematic approach is crucial. The differential diagnosis is extensive and can be categorized to guide clinical reasoning.
Infectious Causes
While bacterial pneumonia was initially suspected in this case, other infections should be considered in the differential diagnosis of bilateral lung infiltrates, including:
- Viral Pneumonias: Influenza, adenovirus, and respiratory syncytial virus (RSV) can present with bilateral infiltrates.
- Fungal Infections: Aspergillus species, Pneumocystis jirovecii (especially in immunocompromised patients), and coccidioidomycosis can cause diffuse lung involvement.
- Mycobacterial Infections: Tuberculosis, although often apical, can present with bilateral disease, especially in disseminated forms.
- Parasitic Infections: While less common in developed countries, parasitic infections like ascariasis and strongyloidiasis can cause eosinophilia and lung infiltrates (Loeffler’s syndrome).
Inflammatory and Autoimmune Conditions
Beyond infections, inflammatory and autoimmune conditions are key considerations when bilateral lung infiltrates and eosinophilia are present:
-
Churg–Strauss Syndrome (Eosinophilic Granulomatosis with Polyangiitis – EGPA): This systemic vasculitis is strongly associated with asthma, eosinophilia, and lung infiltrates. The American College of Rheumatology criteria (Box 1) aids in classification.
-
Hypereosinophilic Syndrome (HES): Defined by persistent eosinophilia and end-organ damage, HES is a diagnosis of exclusion after ruling out secondary and clonal eosinophilia. Diagnostic criteria are shown in Box 2.
-
Allergic Bronchopulmonary Aspergillosis (ABPA): Occurs in patients with asthma or cystic fibrosis, characterized by hypersensitivity to Aspergillus fungi, leading to eosinophilia and lung infiltrates, often with proximal bronchiectasis.
-
Eosinophilic Pneumonia: Can be acute or chronic, characterized by eosinophilic infiltration of the lung parenchyma. Idiopathic eosinophilic pneumonia is diagnosed after excluding other causes.
-
Drug-Induced Lung Disease: Certain medications, including NSAIDs, antibiotics (like nitrofurantoin), and others, can induce eosinophilic lung disease. Montelukast, while less commonly associated, was considered in this case due to its possible link with Churg–Strauss syndrome.
Hematologic Malignancies
Hematologic conditions, particularly myeloproliferative neoplasms, can present with eosinophilia and pulmonary involvement:
- Hypereosinophilic Syndromes (Clonal): These involve underlying genetic abnormalities within the eosinophils themselves, often involving tyrosine kinase function. The F1P1L1-PDGFR-alpha mutation is a key example.
- Chronic Eosinophilic Leukemia, Not Otherwise Specified (CEL-NOS): A rare myeloproliferative neoplasm characterized by persistent eosinophilia without a specific genetic abnormality.
Other Considerations
- Idiopathic Eosinophilic Disorders: When no underlying cause is identified after thorough investigation, the diagnosis may remain idiopathic.
- Loeffler’s Syndrome: A transient pulmonary eosinophilia often associated with parasitic infections but can also be idiopathic.
Evolving Classification and Diagnostic Approach
Historically, eosinophilic disorders were classified based on the site of eosinophilic infiltration or peripheral blood eosinophil counts. However, modern understanding of eosinophilia pathogenesis has led to a simplified approach: determining if the cause is intrinsic (within eosinophils) or extrinsic (outside eosinophil lineage).
Intrinsic disorders are hematologic, often with chromosomal abnormalities affecting tyrosine kinase function and growth factors. Extrinsic disorders are triggered by cytokines, like interleukin-5, stimulating eosinophils or their precursors, often seen in allergic and autoimmune diseases.
Genetic testing for mutations like F1P1L1-PDGFR-alpha is crucial in identifying intrinsic eosinophilic disorders and guiding targeted therapies like imatinib. The absence of such mutations, as in this case, directs clinicians to consider extrinsic or idiopathic causes.
Discussion and Diagnostic Pathway
This case underscores the diagnostic challenges posed by bilateral lung infiltrates and eosinophilia. The patient’s initial presentation resembled community-acquired pneumonia, but the lack of response to antibiotics and the marked eosinophilia prompted a broader differential diagnosis. Her asthma history and pulmonary infiltrates raised suspicion for Churg–Strauss syndrome, while significant eosinophilia suggested hypereosinophilic syndrome. Drug-induced lung disease was also considered given montelukast use.
The diagnostic workup included excluding infections (bacterial, tuberculosis, Legionella), evaluating for vasculitis (Churg–Strauss), and investigating hematologic disorders (hypereosinophilic syndrome). Bronchoscopy and bone marrow biopsy confirmed eosinophilic infiltration, further narrowing the differential. Negative cytogenetic testing for F1P1L1-PDGFR-alpha and normal interleukin-5 levels made clonal hypereosinophilic syndrome less likely, leading to a presumptive diagnosis of Churg–Strauss syndrome initially, later refined to idiopathic hypereosinophilic syndrome after considering the broader clinical picture and response to treatment.
The patient’s favorable response to corticosteroids supported an inflammatory eosinophilic disorder. Hydroxyurea was added for recurrent symptoms, demonstrating the need for tailored treatment strategies in these complex cases. Cardiac involvement, indicated by elevated troponin and echocardiographic findings, highlighted the systemic nature of eosinophilic disorders and the importance of multisystem evaluation.
The evolving classification based on intrinsic versus extrinsic causes of eosinophilia emphasizes the role of genetic and molecular diagnostics. While traditional clinical and pathologic features remain vital, advancements in genetic testing are revolutionizing the diagnosis and management of blood dyscrasias, including eosinophilic disorders. Epigenetic factors also play a role, offering potential therapeutic targets in the future.
Conclusion
Diagnosing bilateral lung infiltrates in the context of eosinophilia requires a comprehensive approach, considering infectious, inflammatory, autoimmune, and hematologic etiologies. This case highlights the complexities and the importance of systematically narrowing the differential diagnosis. While initially resembling common pneumonia, the patient’s clinical course and investigations revealed an underlying eosinophilic disorder. The evolving understanding of these disorders, particularly the distinction between intrinsic and extrinsic causes and the role of genetic markers, is refining diagnostic pathways and therapeutic strategies. For now, idiopathic hypereosinophilic syndrome remains the working diagnosis for this patient, emphasizing the ongoing need for research and advancements in molecular diagnostics to further refine the classification and treatment of these challenging conditions.
Biographies
Christie Lee, MD FRCPC, is a pulmonary and critical care physician at Sunnybrook Health Sciences Centre, University of Toronto, Toronto, Ontario, Canada.
Devon McDonald, MD, is a general internist at Sunnybrook Health Sciences Centre, University of Toronto, Toronto.
Jeannie Callum, MD FRCPC, is a hematologist at Sunnybrook Health Sciences Centre, University of Toronto, Toronto.
Anna Day, MD FRCPC, is professor of medicine, Women’s College Hospital, University of Toronto, Toronto, and a respirologist at the University of Toronto.
Robert Fowler, MD FRCPC, is an internist and critical care physician at Sunnybrook Health Sciences Centre, University of Toronto, Toronto.
Footnotes
Competing interests: None declared. One clinical department to which Robert Fowler belongs has participated in studies sponsored by Astra-Zeneca, Eli Lilly, Wyeth Pharmaceuticals, Novo Nordisk and Chiron Corporation.
Contributors: Christie Lee and Robert Fowler wrote the original draft of the manuscript. All of the authors contributed substantially to the article conception, revised it critically for important intellectual content and gave final approval of the version to be published. Robert Fowler is the guarantor.
References
[1] Masi AT, Hunder GG, Lie JT, et al. The American College of Rheumatology 1990 criteria for the classification of Churg-Strauss syndrome (allergic granulomatosis and angiitis). Arthritis Rheum 1990;33:1094-100. [PubMed]
[2] Cools J, DeAngelo DJ, Gotlib J, et al. FIP1L1-PDGFRalpha fusion kinase in idiopathic eosinophilic syndrome. N Engl J Med 2003;348:1201-14. [PubMed]
[3] Cortes J, Ault P, Koller C, et al. Efficacy of imatinib mesylate in Philadelphia chromosome-positive chronic myelogenous leukemia with resistance or intolerance to interferon-alpha. Cancer Chemother Pharmacol 2000;46 Suppl 2:S99-103. [PubMed]
[4] Mauro MJ, Druker BJ, Maziarz RT. STI571 for chronic myelogenous leukemia. Semin Hematol 2001;38(2 Suppl 2):34-9. [PubMed]
[5] Talpaz M, Silver RT, Druker BJ, et al. Imatinib induces durable hematologic and cytogenetic responses in patients with accelerated phase chronic myeloid leukemia: results of a phase 2 study. Blood 2002;99:1925-31. [PubMed]
[6] Verhoef G, Conneally E, Menke-van der Houven van Oordt C, et al. Imatinib mesylate is effective in patients with chronic eosinophilic leukemia harboring FIP1L1-PDGFRalpha fusion. Haematologica 2006;91:121-3. [PubMed]
[7] Chusid JG, Dale DC, West BC, Wolff SM. The hypereosinophilic syndrome: analysis of fourteen cases with long-term follow-up. Medicine (Baltimore) 1975;54:1-27. [PubMed]
[8] Simon HU, Rothenberg ME, Weller PF. Pathogenesis of eosinophilia. Blood 2010;116:606-17. [PubMed]
[9] Pardanani A, Tefferi A. Hypereosinophilic syndrome and chronic eosinophilic leukemia: 2006 update on diagnosis, risk stratification, and management. Am J Hematol 2006;81:93-117. [PubMed]
[10] Rothenberg ME, Petersen J. Eosinophils in asthma. Chem Immunol Allergy 2007;90:117-41. [PubMed]
[11] Hogan AE, Chouchani ET, Qi NR, et al. Tissue eosinophils are regulators of adipose tissue metabolism. Nat Immunol 2011;12:93-101. [PubMed]
[12] Valent P, Klion AD, Horny HP, et al. Contemporary consensus proposal for diagnosis, risk stratification, and management of hypereosinophilic syndromes and eosinophilic disorders. Blood 2012;120:4298-311. [PubMed]
[13] Tefferi A. Myeloproliferative neoplasms: ASH education book. Hematology Am Soc Hematol Educ Program 2011;2011:485-94. [PubMed]
[14] Gotlib J. World Health Organization-defined eosinophilic disorders: 2011 update on diagnosis, risk stratification, and management. Am J Hematol 2011;86:677-88. [PubMed]
[15] Vardiman JW, Thiele J, Arber DA, et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood 2009;114:937-51.