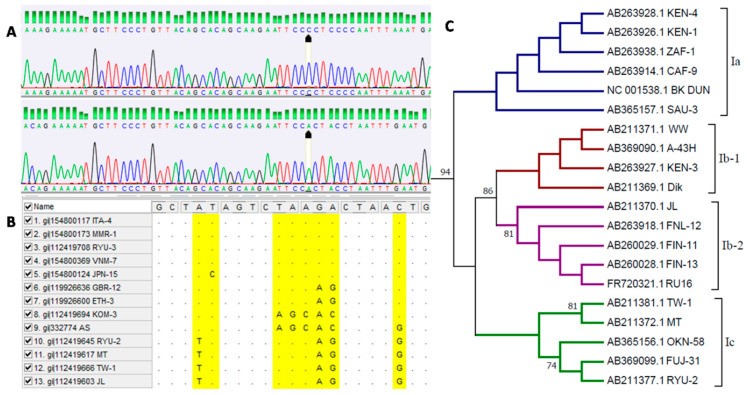
For a comprehensive understanding of BK polyomavirus (BKPyV), especially in the context of effective Bk Diagnosis and treatment strategies, analyzing the distribution of viral variants within populations and determining the evolutionary relationships between different isolates is paramount. Identifying the genotypes of BKPyV, as illustrated in Figure 2, is an essential step in this process. A cornerstone method for bk diagnosis at the molecular level involves sequencing the viral genome.
Figure 2: Bioinformatic analysis of BK polyomavirus sequences crucial for bk diagnosis. (A) Chromatogram analysis representing BKPyV genotypes I and IV. (B) Sequence alignment from databases showing different virus genotypes. (C) Phylogenetic analysis of virus subtypes within genotype I.
The first significant genotyping scheme for BKPyV was introduced by Jin et al. in 1993. This method utilized restriction fragment length polymorphism (RFLP) to detect nucleotide polymorphisms within a short segment of the VP1 capsid protein gene (nucleotides 1744 to 1812). This pioneering work established the concept of genotype variation in BKPyV, initially distinguishing four main genotypes (I–IV). As research progressed and our understanding of BKPyV genetic diversity deepened, sequencing techniques became instrumental in further subdividing these genotypes into subtypes, refining the precision of bk diagnosis.
The complete BK polyomavirus genome was first sequenced and published in 1979. Currently, the NCBI database houses over 4000 nucleotide sequences related to BKPyV. This extensive collection is partly attributable to the rapid advancements in next-generation sequencing (NGS) technologies in recent years. NGS has revolutionized molecular diagnostics, offering enhanced precision in characterizing the viral population within a patient. For instance, Liimatainen et al. (2020) employed NGS to identify and analyze rearrangements in the transcriptional control region (TCR) of BK virus. Furthermore, NGS extends its utility in bk diagnosis to metagenomic analysis, enabling the detection and quantification of various transplant-associated viral infections alongside BKPyV.
Despite the rise of NGS, Sanger sequencing remains a valuable tool for BKPyV genotyping in bk diagnosis. Its continued relevance stems from the relatively short sequence length required for classifying the virus into subtypes. BKPyV genotyping predominantly relies on identifying polymorphisms within the complete VP1 protein-coding sequence. Extensive research focused on this region has elucidated its variability, leading to the development of classification schemes that allow for assigning specific isolates to particular subtypes, as detailed in Table 1.
However, contemporary research is increasingly exploring polymorphisms in other genomic regions, such as the large T antigen and NCCR, to refine BKPyV classification and improve bk diagnosis. Sequencing plays a vital role in identifying BKPyV genotypes circulating within a population, providing critical insights into genotypic variation, global variant distribution, and potential geographic origins. This information is crucial for formulating hypotheses regarding virus evolution and migration, all of which are essential for advancing bk diagnosis and epidemiological understanding. Knowledge of virus genotypes is also indispensable for developing and updating diagnostic assays, studying immune responses to different BKPyV variants, and potentially designing vaccines to stimulate antibody production and improve outcomes for immunocompromised patients infected with BK polyomavirus. Molecular polymorphism, especially within the VP1 protein’s BC loop, can induce genotype-specific antibody synthesis, potentially enabling immune evasion from antibodies targeting other genotypes. Therefore, genotype-specific antibodies are increasingly recognized as important for tailored immunotherapeutic strategies in bk diagnosis and management.
Bioinformatic analysis is also crucial in bk diagnosis, allowing for the evaluation of how nucleotide sequence polymorphisms impact protein amino acid sequences and functionality. Single nucleotide polymorphisms (SNPs) are increasingly recognized for their potential role in viral pathogenesis. Varella et al. (2018) observed variations in viral load between Ia and Ib2 subtypes, which exhibit differences in VP1 and NCCR coding regions, suggesting a link between genotype and disease severity, relevant for bk diagnosis and prognosis. Subtle changes in the nucleotide sequence can influence not only pathogenicity but also viral tropism and host range. In JC polyomavirus, mutations in the VP1 protein’s BC and HI loops have been linked to progressive multifocal leukoencephalopathy (PML). Studies have isolated JCPyV variants with VP1 amino acid sequence mutations from a significant proportion of PML patients. NCCR rearrangements in JCPyV also appear to play a role in PML pathogenesis, with rearranged NCCR variants frequently found in PML cases. These JCPyV variants with rearranged NCCRs exhibit enhanced viral replication and broader host cell susceptibility compared to archetype forms.
Similarly, in BKPyV, heightened molecular variation has been observed in isolates from patients with BKPyV-associated nephropathy (BKPyVAN) compared to healthy individuals or transplant recipients without graft loss. This suggests that viral genetic instability might contribute to immune evasion and antiviral drug resistance, factors that are critically important in bk diagnosis and treatment planning. Luo et al. (2011) also highlighted significant variation in the BK polyomavirus BC loop, leading to quasispecies formation potentially associated with increased pathogenicity. McIlroy et al. noted that in kidney transplant recipients with BKPyV replication, the wild-type variant initially dominates, but rapid replication leads to BC loop mutations in VP1, potentially affecting infectivity and antibody neutralization resistance. Research indicates that BKPyV can manipulate host mechanisms to increase VP1 mutation rates. Verhalen et al. demonstrated that BK polyomavirus can induce expression of the host APOBEC3B gene, while Peretti et al. suggest that the virus can exploit host A3B to acquire mutations that facilitate escape from neutralizing antibodies. Increased molecular variation in BKPyV patients may also result from enhanced replication, increasing the likelihood of severe disease and tissue damage. Prolonged immunosuppression-driven viral replication can lead to mutation accumulation, suggesting that enhanced replication is both a consequence and a driver of higher BKPyV molecular variation, all of which are crucial considerations in bk diagnosis and patient management.
Given the functional significance of polymorphisms, bioinformatic methods are invaluable for predicting and modeling the potential effects of nonsynonymous mutations in the viral genome, as shown in Figure 3. Bioinformatic tools are integrated with databases like NCBI and ENSEMBL, facilitating data exchange and collection on nucleotide and amino acid sequences. Database access minimizes reliance on reference samples, as sequence comparisons against database resources enable definitive pathogen identification and variant comparisons with isolates from diverse global locations. This capability significantly enhances the accuracy and efficiency of bk diagnosis and research efforts.
Figure 3: Prediction of functional effects of mutations in the VP1 protein amino acid sequence using the SNAP2 application, aiding in bk diagnosis. Polymorphic amino acids are shown in relation to the DUN strain. Dark red indicates a strong signal for functional effect (score > 50).
In conclusion, BKPyV genotyping is a cornerstone of modern bk diagnosis, providing essential insights into viral characteristics, evolution, and pathogenesis. Utilizing both traditional and advanced sequencing methods, coupled with powerful bioinformatic tools, allows for a deeper understanding of BKPyV genetic diversity and its clinical implications. This knowledge is critical for improving diagnostic accuracy, guiding therapeutic strategies, and ultimately enhancing patient outcomes in the context of BKPyV infection.