INTRODUCTION
Botulism, a severe paralytic illness, arises from botulinum neurotoxins, potent substances produced by anaerobic, spore-forming bacteria. These bacteria, including Clostridium botulinum groups I and II, Clostridium butyricum, and Clostridium baratii, generate human-pathogenic neurotoxins of types A, B, E, and F. Laboratory diagnostics for botulism are crucial, primarily focusing on detecting botulinum neurotoxin within the patient. Confirmation is achieved by identifying toxin-producing clostridia in the patient or suspected food source. The mouse lethality assay has been the cornerstone of neurotoxin detection. While rapid and sensitive in vitro assays exist, their validation for clinical and food samples is still underway. Culture methods for C. botulinum are underdeveloped, hindering efficient isolation and identification. Molecular techniques targeting neurotoxin genes offer rapid detection of C. botulinum but cannot confirm biologically active neurotoxin presence and shouldn’t be used as standalone diagnostic tools. Beyond immediate diagnosis, botulism laboratory diagnostics plays a vital role in enhancing our understanding of disease epidemiology and informing prevention strategies. Therefore, routine isolation of toxin-producing organisms from patients and suspect vehicles, coupled with physiological group and genetic trait determination, is essential.
Since the late 18th century, with the earliest documented food-borne botulism cases, the disease has become a significant concern. It poses a threat not only to food safety but also as a potential cause of sudden infant death syndrome (SIDS), a risk for intravenous drug users, and even a tool for bioterrorism. Botulism is directly caused by botulinum toxins, exceptionally potent neurotoxins generated during the growth of Clostridium botulinum, a spore-forming bacterium.
C. botulinum strains are categorized into toxinotypes A to G based on the serological properties of their toxins. The taxonomy of C. botulinum is unique, with the unifying feature being the production of neurotoxins causing flaccid paralysis. These strains are classified into four distinct groups (I to IV) based on genotype and phenotype. Groups I and II are primarily associated with human botulism, while group III mainly affects animals, although exceptions occur. Group IV is not generally linked to disease. Group IV C. botulinum has been proposed for reclassification as Clostridium argentinense due to its distinct characteristics. Human-pathogenic strains in group I produce toxins A, B, or F, and group II strains produce toxins B, E, or F. Dual-toxin-producing strains and strains with a single toxin type but carrying silent genes for others have also been identified. Notably, C. butyricum and C. baratii, other clostridial species, can produce type E and F toxins, respectively.
Phenotypically, group I and II C. botulinum strains exhibit significant differences. Group I organisms are more terrestrial and prevalent in temperate climates, while group II strains, particularly type E, are frequently found in colder aquatic environments, especially in the Northern Hemisphere (Table 1). These differences extend to spore heat resistance and growth temperatures, impacting food safety in the industry. Group I spores, highly heat-resistant, are a concern in canned and home-preserved vegetables and meats. Conversely, group II spores, with moderate heat resistance, are a major risk in minimally processed, packaged foods with extended refrigerated shelf lives.
TABLE 1. Characteristics of Clostridium botulinum groups I and II
| Characteristic | C. botulinum group I | C. botulinum group II | Reference(s) |
|---|---|---|---|
| Toxinotypes | A, AB, B, BF, F | B, E, F | 197 |
| Strain origin | Soil | Aquatic environments | 34, 98, 106, 123, 126, 151, 194, 195, 208, 220, 222 |
| Genome size (Mbp) | 3.9-4.1 | 3.6-4.1 | 96, 130 |
| GC content (%) of genome | 26-28 | 26-28 | 115, 127, 128 |
| Genetic diversity | Narrow | Wide | 96, 110, 130, 159, 160 |
| Utilization of proteins as energy source | Yes | No | 35 |
| Carbohydrate fermentation | Glucose, fructose, maltose, salicin, sorbitol | Amygdalin, dextrin, fructose, galactose, glucose, glycogen, maltose, mannose, melezitose, ribose, sorbitol, starch, sucrose, trehalose | 35, 132, 197 |
| Metabolite production | Acetic, butyric, isobutyric, isovaleric, isocaproic, propionic, and valeric acids; alcohols; hydrogen sulfide | Acetic, butyric, formic, succinic, and lactic acids | 197 |
| Bacteriocin production | Boticin | NRa | 51, 125 |
| Colony morphology on blood agar | 2—6 mm (diam); irregular margin; opaque, raised center; narrow hemolysis | 1-3 mm (diam); slightly irregular with lobate margins; translucent to semiopaque with matt surface; mosaic structure in oblique light; narrow hemolysis | 35, 197 |
| Colony morphology on egg yolk agar (lipase reaction) | Iridescent layer on and around colonies | Iridescent layer on and around colonies | 197 |
| Susceptibility to antibioticsb | Susceptible to erythromycin, metronidazole, penicillin, rifampin, tetracycline; resistant to cycloserine, gentamicin, nalidixic acid, sulfamethoxazole, trimethoprim | Susceptible to erythromycin, metronidazole, penicillin, rifampin, chloramphenicol, clindamycin, tetracycline, trimethoprim; resistant to gentamicin, nalidixic acid, cephalotin, sulfamethoxazole | 204 |
| Minimum/optimum growth temp (°C) | 10/35-37 | 3/26-30 | 56, 57, 83, 139, 180, 181, 197, 202 |
| Growth-limiting pH | 4.3-4.5 | 5.0-5.1 | 185, 193 |
| Growth-limiting NaCl in water phase (%) | 10 | 5 | 197 |
| Heat resistance of spores | High (D121-110°C,c 0.15-1.8 min) | Moderate (D85°C,c 1-98 min) | 112, 135, 139, 140, 168–170, 184, 192 |
| Typical vehicle foods for botulism | For food-borne botulism, vegetables, meat, and canned foods; for infant botulism, honey | For food-borne botulism, fish, meat, and minimally processed packaged foods; for infant botulism, NR | 93a, 134 |
aNR, not reported.
bBased on information on C. botulinum type A and E strains (204).
cDT, decimal reduction time at temperature T.
Botulinum neurotoxins are 150 kDa proteins exhibiting zinc-endopeptidase activity. They are secreted as progenitor toxins, comprising the neurotoxin and nontoxic components. These nontoxic components protect the neurotoxin in the environment and aid its absorption into the body. The neurotoxin molecule has two subunits: a 100 kDa heavy chain for binding and translocation across the synaptic membrane via specific receptors, and a 50 kDa light chain. The light chain acts as a zinc-endopeptidase, cleaving proteins essential for acetylcholine vesicle docking and fusion to the presynaptic membrane. This disruption inhibits neurotransmitter release, leading to muscle paralysis. Full toxin potency requires enzymatic activation, possibly involving toxin molecule cleavage. Proteolytic group I C. botulinum strains typically achieve this activation using endogenous enzymes. However, neurotoxins from nonproteolytic group II strains may need external proteases like trypsin for activation.
Botulism is characterized by descending flaccid paralysis. Without prompt and appropriate treatment, it can be fatal due to respiratory muscle failure. Food-borne botulism, the most recognized form, is an intoxication resulting from ingesting preformed botulinum neurotoxin in food. Other forms, like infant botulism, are infections where toxin production stems from spore germination in vivo. Infant botulism occurs when botulinal spores germinate, grow, and produce toxin in the infant’s gastrointestinal tract, where normal gut flora is not fully developed.
Given botulism’s life-threatening nature, rapid diagnosis is critical. This presents challenges for clinical awareness and laboratory diagnostics. Beyond rapid diagnosis and patient survival, understanding botulism epidemiology is crucial for developing prevention strategies. Diagnostics should encompass botulinum toxin and C. botulinum detection, as well as physiological and genetic typing of isolates. The following sections review the current state of laboratory diagnostics for human botulism.
HUMAN BOTULISM
Botulism is a potentially fatal condition caused by botulinum neurotoxin. This neurotoxin can enter the body through the gastrointestinal tract or mucous membranes, such as those in the eyes or respiratory tract. Toxin production can also occur in vivo from spores of otherwise harmless C. botulinum, C. butyricum, or C. baratii. Once in the body, the neurotoxin is absorbed into the bloodstream and lymphatic system, reaching motor nerve endings where it blocks neurotransmitter release. Clinical signs of botulism across all forms include cranial nerve palsies like double vision, dilated pupils, slurred speech, dry mouth, swallowing and speaking difficulties, and facial paralysis. As the disease progresses, limb paralysis and respiratory dysfunction become apparent, potentially leading to death due to respiratory muscle paralysis. Recovery occurs as transitory nerve endings sprout until the original nerve synaptic activity regenerates. Recovery duration can range from weeks to months, depending on the ingested toxin amount and, to a lesser extent, the toxin type. Type A toxin is generally more potent and causes longer-lasting illness compared to types B and E.
Food-Borne Botulism
Food-borne botulism, the classic form, is an intoxication following consumption of food containing preformed neurotoxin. The incubation period can vary from 12 to 72 hours depending on the toxic dose. In addition to general botulism symptoms, gastrointestinal issues such as nausea, vomiting, and constipation may occur. Treatment is primarily intensive symptomatic care, including respiratory support. Specific trivalent antitoxin against toxins A, B, and E can be administered intravenously to neutralize circulating toxin. However, antitoxin effectiveness heavily depends on timely administration; it’s ineffective once the toxin enters nerve endings and clears from circulation. Furthermore, due to the risk of severe allergic reactions to the antiserum, its use is limited or discontinued in some countries.
Differential diagnoses for food-borne botulism include Guillain-Barré syndrome, Miller-Fisher syndrome, chemical intoxication, stroke, and staphylococcal food poisoning. Suspected drug or alcohol abuse can sometimes delay diagnosis.
While food-borne botulism outbreaks are less frequent compared to illnesses or deaths from common pathogens like Campylobacter, Salmonella, and Clostridium perfringens, certain regions have a high incidence, posing significant public health risks. These regions include the Republic of Georgia, Poland, China, Russia, Kyrgyzstan, and specific ethnic groups in northern areas like Alaskan Inuits. Japan, Italy, Portugal, Germany, France, and former Yugoslavia have also reported high case numbers. Frequently implicated foods are home-prepared items such as cured meats, canned vegetables, and fermented fish products. These outbreaks are usually sporadic and family-based. Commercial foods are less commonly involved, but such outbreaks can be large, causing substantial economic losses to the food industry.
The case-fatality rate for food-borne botulism in developed countries ranges from 5 to 10%. Globally, type B toxin accounted for over half of the cases (52%) in 1995, with types A and E contributing 34% and 12%, respectively. Type F toxin is rarely linked to food-borne botulism. Information regarding the physiological group of C. botulinum type B or F strains involved in outbreaks is generally lacking.
Infant Botulism
Unlike food-borne botulism, other human botulism forms are infections with in vivo toxigenesis. Infectious botulism is mainly associated with group I C. botulinum because its optimal growth and toxin production temperature is close to body temperature, while group II growth is limited at this temperature. Infant botulism, affecting babies under one year old, is caused by group I C. botulinum types A and B, Bf, and F. Cases due to type E and F toxins produced by Clostridium butyricum and Clostridium baratii, respectively, have also been reported. The youngest reported infant botulism patient was only 54 hours old. The condition arises from ingesting spores of toxin-producing clostridia. In infants with underdeveloped gut microflora, C. botulinum spores can germinate and establish a toxin-producing culture in the intestine. Clinical manifestations vary from subclinical conditions to sudden death. Constipation lasting several days is often the initial symptom, followed by flaccid paralysis characterized by feeding difficulties due to oral and throat muscle weakness, facial muscle paralysis, ptosis, and general weakness. Infant botulism has been proposed as a potential cause of sudden infant death syndrome (SIDS).
Treatment for infant botulism primarily involves high-quality supportive care, focusing on nutrition and respiratory functions. Antitoxin use is often unnecessary, and the case-fatality rate is less than 2%. However, a human-derived immune globulin available in the United States since 2003 has significantly shortened hospitalization and reduced treatment costs. Honey, potentially containing high C. botulinum spore counts, is the only foodstuff consistently linked to infant botulism. Infant milk powder and environmental dust are also considered potential spore sources.
Wound Botulism
Wound botulism is a rare form, but increasingly diagnosed among injecting drug users due to contaminated needles or impure heroin. It occurs when C. botulinum spores germinate and grow in deep wounds or abscesses providing anaerobic conditions. The clinical presentation mirrors food-borne botulism, but without gastrointestinal symptoms. Wound botulism often involves mixed infections and may be caused by multiple toxin-producing strains. The median incubation period is 7 days. Treatment includes respiratory support, surgical debridement, antibiotics, and antitoxin administration. The estimated case-fatality rate is 15%.
Adult Infectious Botulism
Adult infectious botulism, rare and similar to infant botulism in pathogenesis and clinical presentation, results from intestinal colonization by toxin-producing clostridia. Individuals with altered gut flora due to abdominal surgery, prolonged antibiotic treatment, or gastrointestinal wounds are particularly susceptible. It is distinguished from food-borne botulism by the absence of a history of consuming high-risk foods.
Other Forms of Botulism
Inhalation botulism can result from aerosolized neurotoxin exposure, with a few human cases reported. Iatrogenic botulism, causing localized or generalized weakness, is rare but can occur after therapeutic neurotoxin injections. A notable iatrogenic botulism outbreak in Florida in 2004 was linked to improper botulinum toxin treatment.
LABORATORY DIAGNOSTICS OF BOTULISM
Rapid diagnosis is crucial for successful botulism therapy given its life-threatening nature. Diagnosis relies on clinical presentation, patient history, and positive laboratory findings. Detecting toxin in patient serum or feces remains the gold standard. C. botulinum detection in patient samples (feces, gastric/intestinal contents, wound swabs/tissues) supports diagnosis but is not solely definitive. Multiple toxin-producing strains can complicate diagnostics. In some infant botulism cases, C. botulinum presence in feces or intestinal content, even without detectable neurotoxin, may suffice for diagnosis.
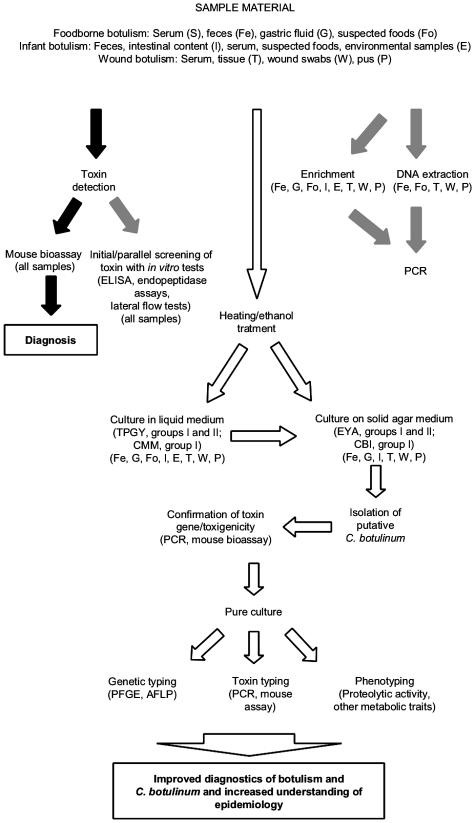
FIG. 1. Laboratory diagnostics of botulism.
 FIG. 1.
FIG. 1.Laboratory diagnostics of botulism. Standard methods are marked with black arrows, and methods aiming at rapid initial screening are marked with gray arrows. Measures aiming at the characterization of the disease isolates and providing complete epidemiological information are marked with white arrows. TPGY, tryptone-peptone-glucose-yeast extract medium; CMM, cooked-meat medium; CBI, C. botulinum isolation medium.
While rapid diagnosis is vital for patient survival, thorough botulism outbreak investigations are necessary for epidemiological data (Fig. 1). This includes C. botulinum isolation from patients and suspect sources, and genotypic analysis of isolates. Characterizing the physiological group of these isolates is fundamental to understanding risk factors and developing prevention strategies for different botulism forms.
The following sections discuss challenges and advancements in laboratory diagnostic tools and procedures for human botulism.
Laboratory Safety
The extreme potency of botulinum neurotoxin necessitates stringent safety protocols for laboratory personnel. Despite its potent toxin, C. botulinum is classified as a class II pathogen (non-invasive, non-contagious). Biosafety level 2 containment facilities and trained staff are minimum requirements. Additional precautions are needed when aerosol or droplet formation from toxic materials is expected. Given the bioterrorism potential of neurotoxins, restricted access to laboratories with toxic cultures is crucial. Contingency measures should be based on thorough risk assessments for each laboratory handling C. botulinum and its toxin. The pentavalent toxoid vaccine previously used for laboratory staff has shown lower than expected efficacy. Therefore, daily safety practices are paramount. Efforts to develop improved botulism vaccines are ongoing in the US and Europe.
Detection of Botulinum Neurotoxin
The mouse lethality assay remains the standard for botulinum neurotoxin detection, but significant progress has been made in developing alternative tests (Table 2). Recent advancements focus on speed and sensitivity, with the fastest tests taking only 20 minutes and surpassing the mouse bioassay in sensitivity. However, further research is needed to achieve rapid, sensitive, single tests capable of detecting all seven neurotoxin types simultaneously. Many new tests are designed for the medical industry, specifically for potency testing of therapeutic botulinum toxin preparations, lacking validation on food or clinical matrices. Complex matrices like feces, blood, pus, and foods can interfere with test reactions. High concentrations of competing microbes in samples can hinder C. botulinum growth and toxin production. Fecal proteinases can degrade botulinum toxin, causing false negatives. Systematic validation of botulinum toxin detection tests in clinical and food samples is essential.
TABLE 2. Diagnostic assays applied in the detection of botulinum neurotoxins
| Assay | Time to perform | Type of toxins | Detection limit | Application(s) | Special feature(s)a | Reference(s) |
|---|---|---|---|---|---|---|
| Mouse lethality assay | 1-4 daysb | A, B, C, D, E, F, G | 20-30 pg/ml, 1 MLD/ml | Bacterial cultures, serum, feces, gastric contents, foods, environmental samples | Standard method | 39, 162 |
| ELISA | 1-2 days | A | 10 MLD50 | Inoculated beans and mushrooms | Polyclonal (horse) Ab as CA | 177 |
| 8 h | A | 4-8 pg/ml, 1-2 MLD/ml | Toxin potency testing in the medical industry | Monoclonal (BA93) and polyclonal (rabbit) Ab as CA | 60 | |
| 8 h | A, B | NRc | Fecal samples related to infant botulism cases | Polyvalent, polyclonal (burro) Ab as CA | 49 | |
| 8 h | A, B | 0.2 ng/ml | Human serum | Polyclonal (horse) Ab as CA | 206 | |
| 8 h | E, F | 0.5-2 ng/ml | Human serum | Polyclonal (horse) Ab as CA | 173 | |
| 8 h | A, B, E | 9-45 pg | Inoculated ground turkey meat | Polyvalent, polyclonal (horse) Ab as CA | 174 | |
| Amplified ELISA | 8 h | A | 5-10 MLD50 | Inoculated canned salmon and corned beef | Monoclonal antibody (BA11), diaphorase-based amplification system | 189 |
| 8 h | A | 10 MLD/ml | Bacterial culture from cheese related to botulism outbreak | Polyclonal (goat) Ab, diaphorase-based amplification system | 66 | |
| 8 h | A, B, E, F | 1-10 MLD, 0.2-1 ng/ml | Chili and potato linked to botulism outbreak | Polyclonal (goat/rabbit) Ab as CA, biotin-streptavidin-based amplification system | 67, 68 | |
| ELISA-ELCA | 5 h | E | Inoculated fish | Polyclonal (chicken) Ab as CA | 178 | |
| 1-2 days | A, B, E | 5-10 pg/ml, 1 MLD | Bacterial cultures | Polyclonal (chicken/horse serum) Ab as CA | 53, 54 | |
| Immuno-PCR | 8 h | A | 5 pg | Purified toxin | Monoclonal antibody (BT57-1), PCR amplification of reporter DNA attached to antibody | 219 |
| Chemiluminescent slot blot immunoassay | 6 h | E | 4 MLD | Bacterial cultures, inoculated fish, naturally contaminated soil | Polyclonal (rabbit) Ab, chemiluminescent detection of slot-blotted culture supernatant | 32 |
| Electro chemiluminescence | 1-3 h | B | Purified toxin | Monoclonal (B 2/3) and polyclonal (horse) Ab, immunomagnetic concentration | 87 | |
| Radioimmunoassay | 8 h | A | 100 MLD50 | Purified toxin | Polyclonal (rabbit) Ab | 23 |
| Lateral flow immunoassays | 15-30 min | A, B, E | 15 pg-10 ng/ml | Inoculated milk products | Immunochromatographic detectiond | 187 |
| 20 min | A | 15-150 pg/ml | Vegetables and seafood | Ganglioside-liposome (GTb1) as CA, immunological detection | 1 | |
| Endopeptidase assay | 8 h | A | 0.1-0.8 MLD50 | Toxin potency testing in the medical industry | Peptide substrate specific for SNAP-25e, immunological detection | 59 |
| 5-6 h | B | 5-10 pg/ml | Inoculated meat pate, cheese, cod, mince, sausages | Immunoconcentration of toxin with monoclonal antibodies, peptide substrate specific for VAMPf, immunological detection | 215, 216 | |
| 8 h | A, B | 0.1-4.5 ng/ml | Bacterial cultures | Peptide substrates specific for VAMP/SNAP-25, immunological detection | 89 | |
| 8 h | A, B, D, F | 2 ng/ml | Toxin potency testing in the medical industry | Fluorigenic peptide substrates specific for VAMP/SNAP-25 | 182 | |
| 8 h | A, B, E, F | 0.04-0.6 MLD50/ml | Purified toxins | Peptide substrates specific for VAMP/SNAP-25, multiplex detection with mass spectrometry | 26 |
aAb, antibody; CA, capture antigen.
bIn 98% of cases a positive result is observed in 24 h (39).
cNR, not reported.
dThe type of antibody was not reported.
eSNAP-25, 25-kDa synaptosomal-associated protein.
fVAMP, vesicle-associated membrane protein.
Mouse Lethality Assay
The mouse lethality assay is the established standard for botulinum toxin detection. This assay involves intraperitoneal injection of diluted samples into mice. Toxin presence is indicated by botulism symptoms in mice, such as ruffled fur, muscle weakness, and a characteristic “wasp-waist” appearance due to respiratory failure. Symptoms typically appear within a day but can take longer. Since nascent toxin requires cleavage for activation, toxins from group II C. botulinum strains need trypsin activation prior to analysis, as these strains lack endogenous proteolytic activity.
Toxin type is determined by neutralization using specific antitoxins. Mice injected with neutralizing antitoxin survive, while others develop botulism. Alternatively, samples can be pre-treated with antitoxins before mouse administration. This method is highly specific and can identify all seven toxin types. The mouse bioassay is very sensitive, with a detection limit of 0.01 ng/ml and one mouse 50% lethal dose (MLD50) corresponding to 5 to 10 pg. It’s used in clinical microbiology labs for feces, serum, gastric, wound, food, and bacterial culture samples. However, the assay is labor-intensive, expensive, raises ethical concerns due to animal use, and is performed in a limited number of labs worldwide. Its turnaround time can also be too slow for timely diagnosis in suspected clinical cases requiring immediate treatment. False positives can occur due to in vivo toxin formation from high C. botulinum spore counts (107), endotoxins from gram-negative bacteria, and tetanus toxin.
Nonlethal Mouse Assay
A nonlethal mouse assay using local muscle paralysis after subcutaneous injection of type A botulinum toxin has been explored for therapeutic potency testing. This assay offers comparable sensitivity and specificity to the traditional bioassay but avoids distress or impaired movement in animals. However, it’s primarily validated for purified neurotoxin potency testing and not for complex microbiological samples.
Immunological Methods
Numerous immunoassay formats for botulinum neurotoxin detection have been developed. Immunoassays offer technical simplicity, speed, and ease of interpretation compared to the mouse test. Early immunoassays like radioimmunoassay, gel diffusion assay, passive hemagglutination assay, and early ELISAs had limited sensitivity or specificity. Recent signal amplification advancements have achieved sensitivities comparable to the mouse bioassay (Table 2). A limitation of immunoassays is the availability of high-quality antibodies. False positives can occur from inactivated toxin, and genetic variation within neurotoxin serotypes can lead to decreased monoclonal antibody affinity, causing false negatives.
ELISA is the most commonly used immunoassay for botulinum neurotoxin detection. In sandwich ELISAs, sample neurotoxin binds to a solid matrix pre-coated with capture antibodies (polyclonal or monoclonal) against one or more toxins. A second toxin antibody then binds the toxin, followed by an enzyme-conjugated anti-antibody to generate a signal via chromogenic substrate cleavage. Conventional ELISA sensitivity is 10 to 100-fold lower than the mouse bioassay. Signal amplification techniques using chromogenic diaphorase systems, biotinylated antibodies with avidin-enzyme conjugates, or enzyme-linked coagulation assays (ELCA) have improved assay sensitivity.
ELISAs have been extensively tested with purified botulinum toxin, toxic C. botulinum cultures, and food samples linked to outbreaks or artificially contaminated. A collaborative study demonstrated high reproducibility of an amplified ELISA with toxin-containing foods. Food components can interfere with ELISA, reducing sensitivity, but successful detection has been reported in fish fillets, canned salmon, corned beef, turkey meat, mascarpone cheese, pasta products, potatoes, chili, and canned beans/mushrooms. Assay limitations should be considered; for example, ELISA-ELCA using chicken and biotinylated antibodies is unsuitable for foods containing chicken, egg yolk, egg white, or milk.
Reports on ELISA performance with clinical specimens like serum and feces are limited. Fecal extracts can significantly interfere with ELISA, reducing sensitivity. Studies in infant botulism cases improved ELISA performance with extended incubation and fetal bovine serum to block interfering fecal substances, showing even greater reliability than the mouse assay, which had false positives. ELISA-ELCA was suggested to perform well with complex clinical matrices like blood and excreta, but systematic validation reports for clinical botulinum toxin detection are lacking. However, ELISA procedures, completed within a workday, are suitable for initial botulism screening (Fig. 1).
A chemiluminescence immunoassay for type E toxin, combined with slot blot transfer of culture supernatants, was reported to be faster than ELISA with relatively high sensitivity. However, false positives occurred with inoculated fish samples. An electrochemiluminescence assay with immunomagnetic concentration of target antigen has sensitivity comparable to the mouse bioassay. Electrochemiluminescence is promising but needs further testing with complex matrices.
Commercial lateral flow assays (dipsticks) offer convenient, rapid toxin testing (15-30 minutes). Assays using the botulinum neurotoxin receptor, trisialoganglioside GT1b, as a capture agent achieved mouse assay-like sensitivity for type A toxin, but fatty foods interfered. Lateral flow assays can be valuable initial screening tools, but negative results, particularly, should be confirmed with the mouse bioassay. Sample pretreatment optimization is crucial to avoid false negatives due to matrix interference.
Endopeptidase Assays
Botulinum neurotoxin’s highly specific zinc-endopeptidase activity on synaptic cleft proteins has inspired in vitro toxin detection assays. Endopeptidase assays detect specific cleavage of synaptic proteins (SNARE complex proteins) by different botulinum neurotoxins, followed by immunological detection of cleaved peptides or fluorescence detection from cleaved quenched-chromophore-labeled peptides.
Endopeptidase assays exist for types A, B, D, E, and F, but commercial solutions are mainly available for type A. Endopeptidase assays have the potential to replace the mouse lethality assay because they detect only biologically active neurotoxin and are generally more sensitive. Mass spectrometry detection of cleaved peptides further enhances sensitivity. However, expensive equipment and specialized skills limit their widespread use. Surface plasmon resonance spectroscopy detection of native SNARE complex protein cleavage by types B and F toxins in rat brain synaptic vesicles achieved the highest sensitivity, 200-fold greater than the mouse assay. This method was considered compatible with complex matrices but lacked reported use for this purpose.
Endopeptidase assays are highly specific, with no cross-reactivity reported between different botulinum toxins or with tetanus toxin. Functional immuno-detection-based assays have successfully detected type B toxin in foods like paté, cod, and cheese. Immunological concentration of neurotoxin using monoclonal antibodies against type B toxin prior to the endopeptidase assay avoided interference from fatty foods. However, false negatives occurred because monoclonal antibodies failed to recognize all type B toxins from different C. botulinum strains.
Further validation is needed for all toxin types, particularly with complex matrices like feces and foods that may contain endogenous proteinases. Substances like EDTA, a common blood anticoagulant, can inhibit type A toxin assays.
Culture Methods for Clostridium botulinum
C. botulinum requires strict anaerobic conditions for growth, posing challenges for laboratory work. Culture media must be deoxygenated by heating or anaerobic gas flow. Reducing agents like thioglycolate can maintain anaerobiosis. Oxygen indicators are essential in media, anaerobic jars, and workstations. Glassware and plastic supplies must be deoxygenated before contacting C. botulinum cultures. Continuous work in an anaerobic workstation is crucial for successful diagnostics.
C. botulinum species comprises physiologically diverse organisms, complicating diagnostics. Conventional detection and isolation involve culturing in liquid media and mouse bioassay detection of toxin in culture supernatants. Positive samples are streaked on solid media, and colony toxin formation is traditionally confirmed by mouse testing. Clinical samples (serum, feces) can be cultured directly or pretreated with ethanol to eliminate vegetative bacteria and recover spores. Heating can also eliminate non-sporeformers, but temperature selection is critical: 80°C for 10 min is recommended for group I spores but may damage group II spores. 60°C for 10-20 min is safer for group II. Adding heat-resistant lysozyme to media can enhance germination of heat-stressed spores.
Routine liquid media include chopped-meat-glucose-starch medium, cooked-meat medium, tryptone-peptone-glucose-yeast extract medium, reinforced clostridial medium, and fastidious anaerobe broth. These are nonselective, allowing growth of various bacteria.
Blood agar and egg yolk agar (EYA) are common unselective plating media. EYA enables lipase reaction, typical of C. botulinum, but many clostridia produce lipase, causing potential misidentification. EYA alone lacks inhibitory compounds, but supplemented with cycloserine, sulfamethoxazole, and trimethoprim, EYA-based selective media (e.g., C. botulinum isolation medium) can select for group I C. botulinum. These media have been used in infant botulism fecal sample investigations. However, they may suppress group II C. botulinum growth, limiting their value in food-borne botulism diagnostics.
Identifying botulinum toxin in/around C. botulinum colonies on agar plates would aid identification and isolation of C. botulinum, C. butyricum, and C. baratii among mixed flora. Immunodiffusion or immunoblotting procedures have been published. A single C. botulinum colony can produce up to 105 minimal lethal doses (MLD) of toxin within 24 hours. Colony immunoblot assays, detecting 10-25 MLD50 of toxin per spot, would be sensitive enough for agar plate identification. However, performance reports with naturally contaminated clinical or food samples or mixed populations are lacking. Fluorescent antibodies against vegetative C. botulinum cell walls have been used for culture identification but suffer from cross-reactivity with nontoxigenic counterparts.
A challenge arises from the different physiologies of group I and II strains in selecting incubation temperature. Group I strains grow optimally at 35-37°C, group II at 25-30°C. A 30°C compromise has been proposed for seeking both groups in a sample. However, replicate sample investigation at 26-30°C and 35-37°C is preferable for optimal growth. Optimal incubation time varies with sample material and detection method. Mouse bioassay for toxin detection in cultures recommends 5-7 days incubation. Shorter incubations may be relevant with molecular techniques like PCR.
Low C. botulinum prevalence in samples (10-1,000 spores/kg) and lack of selective media make plate count quantification difficult in samples with other bacteria. Liquid media enrichment is often needed. Quantification is then done using most-probable-number techniques, based on the amount of positive sample material and the total amount investigated. More culture tubes increase estimate accuracy. C. botulinum presence in culture tubes can be determined by mouse bioassay or PCR.
Discrimination between group I and II strains is essential for epidemiological investigations. Proteolytic activity of group I strains can be determined on casein-containing agar. Molecular techniques like probe hybridization or ribotyping can also differentiate groups. Although group I and II strains differ, both can grow at 30-37°C, a commonly used range. Temperature growth ability should not solely define physiological group.
Commercial biochemical reaction test systems exist for anaerobic bacteria identification. Conflicting reports on their Clostridium spp. identification ability exist, with correct species-level identification ranging from 54% to 96%. Diverse C. botulinum physiology hinders species-level identification for all strains. Biochemical test systems are also susceptible to technical bias, with incubation environment, time, and cell suspension concentration affecting identification success.
Identification and isolation of C. botulinum from samples with high competitive flora, like fecal and environmental samples, are laborious and time-consuming. Multiple broth cultures and platings are needed for pure culture isolation, often failing. Nontoxigenic C. botulinum-like strains can interfere with C. botulinum culture. Whether these are distinct species or derived from toxigenic C. botulinum via toxin gene cluster deletion during subculture needs further investigation. In type E and F toxin botulism, toxin-producing C. butyricum or C. baratii should be considered unless C. botulinum is identified, as these bacteria differ physiologically from C. botulinum, and C. botulinum laboratory routines are not directly applicable.
Molecular Detection of Clostridium botulinum
DNA-based detection methods, particularly PCR and Southern hybridization, have become prominent for C. botulinum detection due to their sensitivity, specificity, and speed compared to culture and mouse bioassays (Table 3). Many labs are equipped and experienced with molecular techniques. Molecular detection in C. botulinum primarily focuses on detecting the botulinum neurotoxin gene (bot) in samples, not necessarily gene activity or toxin presence. While this is a limitation, molecular techniques avoid animal use and are suitable for screening bacterial colonies, pure cultures, and sample enrichments for neurotoxin genes, indicating C. botulinum and other toxin-producing clostridia presence.
TABLE 3. PCR and probe hybridization protocols applied for the detection and identification of Clostridium botulinum groups I and II in different types of samples
| Sample type and pretreatment | Assay type | Toxin types detected | Sensitivity | Reference(s) |
|---|---|---|---|---|
| Extracted DNA from pure cultures | PCR + gel electrophoresis | A, B, E, F | 0.3 ng | 42 |
| PCR + gel electrophoresis | A, B, C, D, E, F | 2.5 pg | 207 | |
| PCR + probe hybridization | A | 12.5 fg | 62 | |
| Real-time PCR | A, B, E | 42-195 fg | 2 | |
| Real-time PCR | A | 0.1 ng | 223 | |
| Multiplex PCR + hybridization onto array | A, B, E, F | 0.015-0.5 pg | 78 | |
| Extracted DNA from spores inoculated in foods | Real-time PCR | A | 102-103 spores/ml | 223 |
| Extracted DNA from enriched foods | PCR + probe hybridization | A, B, E, F | 0.1 cell/g | 8 |
| enriched foods | PCR + probe hybridization | A, B, E, F, G | 10 cells/g | 61 |
| PCR + probe hybridization | A, B, E | 0.1-21 spores/g | 27 | |
| PCR + capillary electrophoresis | E | 10 cells | 183 | |
| Extracted DNA from enriched foods, environmental samples, and clinical samples | PCR + gel electrophoresis | A, B, E | 102 spores/g | 205 |
| Extracted DNA from enriched feces | Nested PCR + gel electrophoresis | B | NRa | 117 |
| Extracted DNA from enriched foods and environmental samples | PCR-ELISA | A, B, E, F | 10−1 cell/g | 63 |
| Extracted DNA from heated and enriched feces | Nested PCR + gel electrophoresis | B, E, F | 10-103 spores/g | 43, 44 |
| Extracted DNA from foods and feces | Real-time PCR | A, B, E | NR | 2 |
| Extracted DNA from foods | Multiplex PCR + gel electrophoresis | A, B, E, F | NR | Wyatt et al., Abstr. Rapid Methods 2005 |
| Extracted DNA from wound swabs, tissues, pus | Real-time PCR | A, B, E | 10-102 cells | 3 |
| Crude cell lysate from | PCR + probe hybridization | A, B, E | NR | 73 |
| enrichment broth | Multiplex PCR + gel electrophoresis | A, B, E, F | 10-102 cells | 133 |
| Multiplex PCR + hybridization onto array | A, B, E, F | 4 × 102 cells | 78 | |
| Crude cell lysate from enriched foods | PCR + probe hybridization | A | 10-103 cells/g | 62 |
| Crude cell lysate from enriched foods and feces | Multiplex PCR + gel electrophoresis | A, B, E, F | Food, 10−2-10−1 spore/g; feces, 10−1-103 spores/g | 133 |
aNR, not reported.
Most PCR-based C. botulinum detection reports focus on single-toxin-type gene detection per reaction. Multiplex PCR assays, enabling simultaneous detection of botA, botB, botE, and botF genes in one reaction, are an improvement, reducing assay time, labor, and reagent costs. Multiplex PCR products are detected using gel electrophoresis or hybridization onto cDNA probe-coated membranes, yielding similar sensitivities.
Molecular detection based on botB and botF genes, highly similar in group I and II C. botulinum, doesn’t differentiate physiological groups. A 23S rRNA gene-specific oligonucleotide probe detects group II strains, but it also recognizes nontoxigenic C. botulinum-like strains within group II, limiting its sole diagnostic value. A group I C. botulinum-specific HindIII fragment-based DNA probe, not hybridizing to C. sporogenes, is more suitable. Molecular typing methods like ribotyping and amplified fragment length polymorphism (AFLP) can also differentiate C. botulinum groups I and II.
Reported PCR and probe hybridization assay sensitivities for C. botulinum detection in feces, serum, and food samples vary significantly. Extracted DNA templates generally yield greater sensitivity than crude cell lysates. However, DNA extraction can be laborious, while cell lysates are easily prepared within an hour.
Nested PCR, using two primer sets for the same gene in sequential reactions, can enhance PCR sensitivity. Nested PCR protocols exist for botB, botE, and botF genes. Nested PCR may shorten or eliminate enrichment steps often needed for standard PCR. Type B C. botulinum was directly detected from infant botulism feces using nested PCR. However, assay detection limits need to be determined before reliable diagnostic application.
Components in clinical and food samples, such as bile salts or competitive flora in feces, blood immunoglobulins, and high protein/fat content in foods, can inhibit PCR or reduce sensitivity. For example, C. botulinum type E PCR detection limit in feces was 4 log units higher than in fish or meat. This must be considered when establishing PCR protocols for clinical specimens. Each sample type requires PCR optimization and sample preparation, focusing on concentrating target cells, eliminating competitive flora, and removing PCR inhibitors. Internal amplification controls and other controls should be routinely used in diagnostic PCR to manage inhibition and contamination. Reports of their use in C. botulinum molecular diagnostics are still limited.
Low C. botulinum organism numbers in naturally contaminated samples and spore formation often lead to PCR failure directly from samples due to insufficient sensitivity. Enrichment is often needed to germinate spores and increase target cell concentration. For samples potentially containing both group I and II C. botulinum, enrichment conditions must be carefully considered, ideally using replicate samples incubated at both 35-37°C (group I) and 26-30°C (group II). Enrichment duration for optimal PCR performance is also crucial and should be determined for each sample material and enrichment medium. Too short enrichment may allow competitive bacteria to overtake C. botulinum, while excessive incubation may cause lysis or sporulation. Ideally, PCR is performed on cultures over several days to determine optimal enrichment length for each sample type.
Direct sample PCR analysis can also detect dead cells due to intact DNA post-lysis. Enrichment procedures partly address this by increasing live cell proportion. Reverse-transcription PCR (RT-PCR), detecting gene expression instead of the gene itself, can ensure only live cells are detected. RT-PCR protocols exist for botB and botE genes. However, in botulism outbreaks, laborious high-quality RNA extraction may be too time-consuming.
Real-time PCR, using different chemistries (intercalating dyes or quencher probes), allows real-time amplification monitoring and rapid result interpretation. Amplicon melting temperature can also be calculated, differentiating specific PCR products from nonspecific amplicons. Real-time PCR has been used for qualitative detection of C. botulinum types A, B, and E in foods and feces from food-borne and infant botulism cases. It has also been applied to wound botulism samples (tissue, wound swabs, pus). Direct DNA extraction and PCR of tissue samples from wound botulism cases showed limited positivity. PCR inhibition was suggested for some negative results. However, after 1-5 days of liquid enrichment, most samples from patients with serum toxin detection showed neurotoxin gene presence.
Beyond speed, real-time PCR can quantify target sequences, reflecting organism levels. Reliable quantification requires knowing target gene copy number in the organism’s genome. Reagent selection, reaction standardization, and sample pretreatment are more stringent than for conventional PCR. Quantitative botB gene detection has been used for research. Recent applications include quantitative detection of type A C. botulinum spores spiked in canned corn and sausage. This method directly extracts DNA from spores, skipping enrichment, allowing same-day analysis. However, speed comes at the cost of sensitivity, detecting no fewer than 100 spores. Natural C. botulinum contamination levels in samples (10-1,000 spores/kg) often necessitate enrichment for successful detection.
As mentioned, C. botulinum strains with silent toxin genes can cause false positives in PCR. Conversely, point mutations or genetic variation at primer sites can cause false negatives, even with functional genes. Therefore, while PCR is a powerful botulism diagnostic screening tool, results should ideally be confirmed by other methods. PCR specificity should be confirmed by combining PCR with bot gene-specific probe hybridization or DNA sequencing of PCR products, instead of or in addition to gel electrophoresis. Sole reliance on PCR results is risky and should be avoided if possible. It’s also important to remember that PCR does not directly detect botulinum neurotoxin.
Genetic Characterization of Clostridium botulinum
Various molecular typing tools are used for C. botulinum genetic characterization, including DNA sequencing, pulsed-field gel electrophoresis (PFGE), ribotyping, and PCR-based techniques like AFLP, RAPD, and Rep-PCR (Table 4). International molecular typing libraries with genetic fingerprints, ideally using multiple techniques, of C. botulinum strains from botulism outbreaks would be valuable for investigating new outbreaks. The modern food industry’s focus on long shelf life and refrigerated storage can lead to widespread product distribution, potentially causing geographically dispersed botulism outbreaks that are challenging to trace and resolve.
TABLE 4. Genetic typing tools applied for Clostridium botulinum groups I and II
| Method | Discrimination of groups I and II (level) | Effort and time required to completion from pure colony | Reproducibility | Special feature | Application(s) | Reference(s) |
|---|---|---|---|---|---|---|
| PFGE | Strain | Laborious, 5-7 days | Excellent | DNA degradation by clostridial endonucleases may disturb analysis | Epidemiological studies, comparison of clinical isolates | 96, 110, 130, 131, 160, 161 |
| rRNA gene sequencing | Group | Moderate, 3-4 days | Excellent | Taxonomy | 33, 41, 107, 108 | |
| Ribotyping | Group | Laborious, 3-5 days (for automated ribotyping, easy, 1 day) | Moderate | Can be automated (RiboPrinter) | Phylogeny, species/group identification | 97, 191 |
| AFLP | Strain and group | Easy, 3 days | Excellent | Epidemiological studies, comparison of clinical isolates, phylogeny | 120 | |
| RAPD | Strain | Easy, 1-2 days | Low | Can be performed with crude cell lysate (although this further reduces reproducibility) | Rapid comparison of clinical isolates, not suited to interlaboratory comparison of strains | 109 |
| Rep-PCR | Group I, toxinotype; group II, strain | Easy, 2 days | Moderate | Not well suited to gram-positive bacteria | Rapid comparison of clinical isolates | 109 |
Pulsed-Field Gel Electrophoresis (PFGE)
PFGE macrorestriction pattern analysis is a powerful genetic typing tool for foodborne pathogen diagnostics. It involves electrophoresis of genomic DNA digested with rare-cutting restriction enzymes, generating a unique fingerprint for each strain, typically with 5-15 fragments (10-1,000 kbp). PFGE can trace botulism outbreak sources by genetically comparing C. botulinum isolates from patients and suspect foods. It’s also used to study C. botulinum biodiversity.
Early PFGE genotyping of group I A toxin-producing strains used MluI, RsrII, and SmaI enzymes. Different strains showed distinct PFGE patterns, while isolates of the same strain had similar patterns. Recent studies recommend SacII, alone or with SmaI and XhoI, for group I PFGE typing. Analysis of 55 group I type A, B, AB, and F organisms revealed high similarity within types AB and B, and type F organisms. For group II C. botulinum, XhoI and SmaI PFGE proved highly discriminative, showing significant genetic diversity among strains from aquatic environments.
While reproducible and discriminative, PFGE is labor-intensive and takes several days. It’s also sensitive to DNA degradation by extracellular DNases, and not all strains, particularly group II, are typeable with standard procedures. Formaldehyde cell fixation has resolved this issue in most cases.
Ribotyping
Conservative ribosomal genes (rRNA genes) are extensively used for phylogenetic analysis. Bacterial rRNA gene restriction pattern analysis (ribotyping) uses genomic restriction patterns hybridized with labeled E. coli rRNA gene cDNA probes. Ribotyping has lower discriminatory power than PFGE for strain-to-strain differentiation but sufficient power to differentiate species, making it ideal for phylogenetic analysis. It effectively discriminated between 8 group I and 19 group II C. botulinum strains using EcoRI, confirming distinct phylogenetic lineages. Ribotyping is highly reproducible, but DNA degradation can hinder typing of some strains. RiboPrinter, an automated ribotyping system, is also used for C. botulinum.
Amplified Fragment Length Polymorphism (AFLP)
AFLP is a relatively new genetic typing technique involving digestion of genomic DNA with two restriction enzymes, adapter ligation, and PCR amplification of fragment subsets. AFLP analysis of 33 group I and 37 group II C. botulinum strains with HindIII and HpyCH4IV showed it to be fast, reproducible, highly discriminative, and excellent for C. botulinum typing. AFLP, like ribotyping, is suitable for phylogenetic analysis and effectively distinguishes group I and II strains. AFLP is not affected by DNA degradation, providing 100% typeability.
Randomly Amplified Polymorphic DNA Analysis (RAPD)
RAPD, also known as arbitrarily primed PCR, uses PCR with random universal primers under low stringency. It’s fast and easy, and reported to be more discriminatory than PFGE for group II C. botulinum. Discriminatory power is lower for group I strains. All strains are typeable with RAPD. However, due to random primer annealing, RAPD lacks reproducibility and is not well-suited for interlaboratory strain comparison.
Repetitive Element Sequence-Based PCR (Rep-PCR)
Bacterial genomes contain conserved repetitive extragenic elements that can be targeted by PCR with consensus primers. Amplification product number and size define a species-specific fingerprint, but strain-level differentiation is limited, restricting its epidemiological and clinical research applicability. A Rep-PCR protocol for C. botulinum differentiated group II type B and E toxin-producing strains to strain level, while group II type F and all group I strains were differentiated only to toxinotype level. Group-specific amplification fragments were obtained for both groups, suggesting Rep-PCR could determine physiological group in botulism cases. Rep-PCR is rapid and more reproducible than RAPD due to specific primer use.
Findings from different genetic typing tools suggest group I and II strains may have different environmental epidemiology, with narrower diversity in group I. This could be due to different life cycles in soil and water; aquatic environments may be more conducive to frequent spore germination, leading to greater genetic variation in aquatic organisms like group II C. botulinum. Whether this is true or typing tools are insufficiently discriminatory for group I requires further investigation.
FUTURE PERSPECTIVES
Significant progress has been made in in vitro botulinum neurotoxin detection tests potentially replacing the mouse assay, and in molecular tools for C. botulinum detection and characterization. However, further advancements are needed. In vitro toxin tests require improved speed and sensitivity in single tests, ideally detecting all seven toxin types simultaneously. These tests also need validation across complex matrices (feces, foods, environmental samples), beyond purified toxins.
While molecular C. botulinum detection and identification methods are promising, conventional culture techniques remain important. Selective media effective for both group I and II C. botulinum and other toxin-producing clostridia are needed to isolate all potential toxin-producing strains from samples with competitive bacteria. The distinct physiologies of C. botulinum groups complicate detection and isolation, suggesting a need to reconsider species taxonomy to better reflect genetic variation among neurotoxin-producing strains.
While rapid laboratory diagnosis is crucial for patient care, comprehensive laboratory procedures should aim for a deeper understanding of botulism epidemiology, risk factors, and prevention, considering both C. botulinum groups. Isolation and genetic characterization of C. botulinum from patients and sources should be pursued whenever feasible (Fig. 1).
Bacterial genome sequencing has opened new possibilities in genetic typing and research into genetic mechanisms underlying metabolic traits like neurotoxin formation and regulation. DNA microarrays enable genome-wide microorganism analysis and may soon supersede conventional genetic typing. While bacterial genomes are being sequenced rapidly, more genomic information on closely related species and strains is needed. The genome of a group I C. botulinum strain (type A, ATCC 3502) has been sequenced, enabling C. botulinum microarray development. However, differences between groups I and II may limit type A array applicability to group II analysis. Sequencing a group II C. botulinum genome is necessary. Genomic information from a single strain is insufficient for a complete understanding of species genetic diversity. Increased research funding for C. botulinum genome sequencing projects is needed to illuminate genetic diversity and mechanisms behind metabolic phenomena in botulinum neurotoxin-producing clostridia.
CONCLUSIONS
Rapid diagnosis is crucial for managing potentially lethal botulism, necessitating active assay development. Rapid, sensitive in vitro assays for detecting all botulinum toxin types are needed, along with thorough validation for complex matrices. Molecular detection and identification tools should be used in screening and surveillance to reduce mouse bioassay use. More sophisticated and efficient tools are needed to isolate group I and II strains from various samples. To improve culture methods, C. botulinum taxonomy should be reconsidered to align with genetic variation among neurotoxin-producing bacteria.
While rapid laboratory diagnosis focuses on patient toxin detection, thorough epidemiological investigation of outbreaks and causative organisms is essential for understanding and preventing different botulism forms. Laboratory diagnostics for each human botulism case should include attempts to isolate C. botulinum from patients and sources. In addition to toxin type identification, isolates should be typed to reveal physiological group. Genetic characterization of isolates should be incorporated into routine diagnostics and epidemiological investigations (Fig. 1).
Acknowledgments
This work was supported by the Academy of Finland (grants 206319 and 207330).