History of Plague and its Potential as a Weapon of Bioterrorism
Pandemic History and Epidemic Potential
Plague, a disease that has shaped human history, has been responsible for over 200 million deaths across three major pandemics in the last two millennia, causing immense social and economic upheaval (Perry and Fetherston, 1997; Pollitzer, 1954). The first, the Justinian Plague, emerged in North Africa in the 6th century and spread throughout the Mediterranean and Near East by the 7th century, significantly weakening the Roman and Byzantine Empires. The second, the Black Death, began in Central Asia, reaching Sicily in 1347 via Crimean ships, and swiftly decimated medieval Europe. By 1352, it had wiped out at least 30% of Europe’s population, with recurring epidemics for centuries, including the Great Plague of London in 1665 (Perry and Fetherston, 1997). The third, or Modern Pandemic, originated in southwestern China in the mid-19th century, hitting Hong Kong in 1894 and spreading globally via steamships to port cities, including those in the US (Link, 1955; Pollitzer, 1954). This pandemic resulted in over 26 million cases and 12 million deaths by 1930. Predominantly bubonic, these pandemics also featured terrifying outbreaks of pneumonic plague, a highly contagious and deadly form spread person-to-person. The Manchurian epidemics of the early 20th century starkly illustrated pneumonic plague’s severity, with tens of thousands of cases and near-universal fatality (Wu, 1926).
Improved sanitation, hygiene, and advanced disease control have significantly reduced plague’s public health impact since the 20th century, with around 2,500 cases reported annually worldwide (World Health Organization, 2003). However, Yersinia pestis, the bacterium causing plague, persists in rodent populations across all continents except Australia (Gage, 1998; Gratz, 1999b). Eradicating these natural reservoirs is practically impossible. While antibiotics have lowered bubonic plague’s fatality rate to 10% or less, pneumonic plague remains highly lethal. A US study of 420 plague cases (1949–2000) found 55 pneumonic plague cases with a 40% fatality rate (Centers for Disease Control and Prevention, unpublished data). For primary pneumonic cases, the fatality rate was even higher at 57.1% (Centers for Disease Control and Prevention, 1997). Although large-scale pandemics are unlikely now, plague, particularly pneumonic plague, retains significant epidemic potential (Boisier et al., 2002; Campbell and Hughes, 1995; Chanteau et al., 1998, 2000; Gabastou et al., 2000; Ratsitorahina, 2000a). This potential has raised concerns about its exploitation for bioterrorism and warfare.
Plague as a Weapon of Biological Warfare
The concept of plague as a weapon dates back centuries. Historical accounts describe the use of plague-infected corpses catapulted into enemy territories in the 14th and 18th centuries (Derbes, 1996; Gasquet, 1908; Marty, 2001). During World War II, the Japanese military conducted unethical plague experiments on humans in Manchuria and disseminated Y. pestis-infected fleas over Chinese civilian areas, resulting in localized bubonic plague outbreaks and rat infections (Bellamy and Freedman, 2001; Harris, 1992; Kahn, 2002). The Cold War saw intensified biological warfare research by the Soviet Union (USSR) and the US, both of which had programs to weaponize Y. pestis by the 1960s. In 1970, a WHO expert committee highlighted plague’s dangers as a weapon due to its high infectivity, ease of mass production and storage, and potential for aerosol dispersal (World Health Organization, 1970). Models predicted that aerosolizing 50 kg of Y. pestis over a city of 5 million could cause 150,000 pneumonic plague cases and 36,000 deaths initially. Without proper measures, a pneumonic plague outbreak affecting 50% of the population could infect 90% within a month, with a 60–70% fatality rate. This report contributed to the 1972 Biological Weapons Convention, effective in 1975, prohibiting biological weapon development (Marty et al., 2001).
Despite signing the convention, the USSR secretly continued an aggressive bioweapons program, stockpiling plague weapons (Alibek, 1999). They developed aerosolized Y. pestis in liquid and dry forms for bomblet deployment, considering plague a strategic weapon. They also engineered virulent, drug-resistant Y. pestis strains to evade vaccines and antibiotics, and had the capacity to produce hundreds of tons of plague weapons (Alibek, 1999). The US military also recognized aerosolized Y. pestis‘s potential for large-scale attacks (Martin and Marty, 2001; Marty, 2001), but their weaponization efforts were less successful due to production challenges. The US halted offensive bioweapons research in 1970, but Soviet programs continued until at least 1990. While Russia has converted civilian facilities, the status of their military program and weapon stockpiles remains uncertain (Alibek, 1999). Many nations maintain defensive bioweapons programs, but the line between defense and offense can be blurred, particularly in studies enhancing understanding of potential agents and their deployment.
US Countermeasures to Plague as a Weapon of Terrorism
Biological weapons development is no longer limited to advanced nations, extending to rogue states, terrorist groups, cults, and individuals. Responding to the growing terrorist threat, the US Congress enacted the Biological Weapons Act of 1989 and the Chemical and Biological Weapons Control and Warfare Elimination Act of 1991 (Ferguson, 1997). Following terrorist attacks like the World Trade Center and Oklahoma City bombings, and bioweapon hoaxes, the Anti-Terrorism Act of 1996 was passed (Anonymous, 1996; Ferguson, 1997). This act directed the CDC to identify dangerous biological agents for terrorist use (select agents) and regulate their acquisition, use, and transfer (U.S. Department of Health and Human Services, 1996). Y. pestis was designated a Category A select agent, posing the highest national security risk (Centers for Disease Control and Prevention, 2000, 2002; Rotz et al., 2002). Since 1997, federal law (42 FR 72.6) (Anonymous, 1997) has strengthened Y. pestis transfer regulations, requiring official transfer forms between facilities. These rules were augmented by 42 CFR part 73 (Possession, Use, Transfer of Select Agents and Toxins) under the 2002 Public Health Security and Bioterrorism Preparedness and Response Act (effective 2003). Part 73 outlines requirements for laboratories handling plague and other select agents, including registration, security assessments, safety, security, and emergency response plans, training, record keeping, inspections, and notifications (Centers for Disease Control and Prevention, 2002). These regulations have sparked debates about balancing scientific freedom and civil liberties with counterterrorism efforts (Annas, 2002; Fidler, 2001).
Preparedness and Response to a Possible Plague Attack
The CDC has developed a strategic plan for responding to the deliberate release of plague and other select agents (Centers for Disease Control and Prevention, 2000; Khan et al., 2000; Rotz et al., 2002). The Johns Hopkins University formed a Working Group on Civilian Biodefense Strategies, creating consensus recommendations for responding to biological weapons attacks using plague or other agents (Johns Hopkins Center for Civilian Biodefense Strategies, 2003; Inglesby et al., 2000). A key outcome was the Model State Emergency Health Powers Act, providing a framework for state officials to assume necessary powers to detect and contain catastrophic disease outbreaks, ranging from pre-emergency planning to property compensation, including disease tracking, property management, and personal protection (Johns Hopkins Center for Civilian Biodefense Strategies, 2003; Mair et al., 2002).
A terrorist attack using Y. pestis aerosol is considered most likely, potentially causing numerous severe pneumonic plague cases. Plague’s notoriety alone could trigger public panic, generate a “worried-well” population, irrational behavior, and overwhelm medical and emergency services (Glass and Schoch-Spana, 2002; Osterholm and Schwartz, 2000; O’Toole and Inglesby, 2001). This scenario was evident during the 1994 plague emergency in Surat, India (Dennis, 1994; Ramalingaswami, 2001). US bioterrorism preparedness includes stockpiling drugs, vaccines, and equipment; establishing emergency operations centers with advanced communications; and a CDC terrorism response unit working with the Department of Homeland Security (Centers for Disease Control and Prevention, 2000; Centers for Disease Control and Prevention, Bioterrorism Program, 2003; Rotz et al., 2002). Plague attack simulations like TOPOFF I (Denver, 2001) and TOPOFF II (Chicago, 2003) have identified critical emergency response gaps, including leadership and decision-making issues, resource prioritization, information sharing failures, and overwhelmed healthcare facilities (Block, 2003; Hoffman, 2003; Hoffman and Norton, 2000; Khan and Ashford, 2001; Inglesby et al., 2001). These simulations highlighted the urgent need for pre-defined disease containment principles and the legal authority to implement them effectively.
Plague Microbiology and Pathogenesis
The Agent
General Characteristics
Yersinia pestis is a Gram-negative, nonmotile, non-sporulating, microaerophilic, pleomorphic coccobacillus (1.0-2.0 μm × 0.5 μm) belonging to the Enterobacteriaceae family (Perry and Fetherston, 1997). In smears, it appears as single cells or short chains, characteristically exhibiting bipolar staining (safety-pin appearance) with Wayson, Giemsa, or Wright stains. It is catalase-positive, and urease-, oxidase-, and indole-negative. Y. pestis is biochemically relatively inert, potentially leading to misidentification by automated systems if not properly configured (Wilmoth et al., 1996). It grows in various media at temperatures from 4 to 40°C (optimal 28-37°C) and pH 5.0-9.6 (optimal 6.8-7.6). Y. pestis grown at 28-30°C and pH over 7.2 is more stable in the environment and in aerosol form (K. Alibek, personal communication). It is slow-growing, with pinpoint colonies on sheep blood agar visible after 24 hours at 28°C, and later on MacConkey agar. Colonies are raised, opalescent, and develop a hammered copper surface with irregular borders as they enlarge. In broth, a stalactite-like growth pattern forms along the vessel sides, settling in clumps if disturbed. Most Y. pestis strains are susceptible in vitro to tetracyclines, chloramphenicol, sulfonamides, aminoglycosides, and fluoroquinolones (Frean et al., 1996, 2003; Smith et al., 1995). Rare isolates with incomplete susceptibility to recommended antimicrobials have been found, but these have not led to widespread resistance. However, the 1995 identification of a multidrug-resistant Y. pestis strain in Madagascar, with plasmid-mediated and transferable resistance (Galimand et al., 1997), raised significant concern and prompted calls for increased surveillance (Dennis and Hughes, 1997). Fortunately, further spread of such strains has not been detected.
Molecular Genetics
Genetic analysis indicates Y. pestis evolved recently (1,500–20,000 years ago) from Y. pseudotuberculosis, an enteric pathogen (Achtman et al. 1999). Genome sequencing of Y. pestis (4.65 Mb chromosome and three plasmids) revealed its evolution involved acquiring virulence factors for systemic invasion and flea replication, and losing genes for enteric survival (Parkhill et al., 2001). These genomic changes highlight the dynamic evolution of highly virulent pathogens. Three classic Y. pestis biovars are recognized: Antiqua, Medievalis, and Orientalis, linked to the three historical pandemics. Ribotyping supports these biovars, showing chromosomal rearrangements in the Orientalis biotype after its global spread about 100 years ago (Guiyoule et al., 1994). Polymorphism studies reveal considerable genome plasticity, even within strains from the same geographic area (Guiyoule et al., 1997; Radnedge et al., 2002).
Pathogenicity of Y. pestis
Virulence Factors
Y. pestis is exceptionally pathogenic. While a facultative intracellular pathogen, its virulence depends partly on invading and multiplying within cells, including phagocytes that facilitate early infection stages (Hinnebusch, 1997; Perry and Fetherston, 1997). Both chromosomal and plasmid-encoded genes contribute to host adaptation and virulence (Carniel, 2003; Hinnebusch, 1997; Hinnebusch et al., 2002; Koornhof et al., 1999; Perry and Fetherston, 1997; Smego et al., 1999). Chromosomal genes express a potent lipopolysaccharide endotoxin and a factor controlling exogenous iron absorption. The three major plasmids include: the pesticin (pst) plasmid (~9.5 kb) encoding plasminogen activator (Pla) and pesticin (Pst); the low calcium response (Lcr) plasmid (~70 kb) encoding V and W antigens and outer surface proteins (Yops); and the pFra (Tox) plasmid (~110 kb) encoding F1 glycoprotein envelope antigen (Caf1) and murine toxin (Ymt). F1 antigen, produced at temperatures ≥30°C, aids resistance to phagocytosis in the absence of opsonizing antibodies.
Y. pestis virulence factors mediate the following host-pathogen interactions:
- Lipopolysaccharide endotoxin: Activates complement and releases proinflammatory mediators.
- Hemin storage molecule (Hms): Enhances Y. pestis survival in phagocytes and uptake into eukaryotic cells.
- pH 6 antigen (Psa): Inhibits Y. pestis phagocytosis.
- Plasminogen activator (Pla): Degrades fibrin and extracellular proteins, facilitating systemic spread.
- V and W antigens: Block phagocytosis, with V antigen promoting survival in macrophages.
- Yops: Inhibit phagocytosis and platelet aggregation, and suppress inflammatory responses.
- F1 antigen: Antiphagocytic and elicits a strong humoral immune response.
Factors selectively expressed in the flea gut, like hemin storage locus (hms) products, protect Y. pestis from cytotoxic digestion by blood plasma products (Perry, 2003).
Pathology of Infection
Y. pestis virulence manifests in a range of diseases based on entry portal and targeted organs (Butler, 1972, 1983; Crook and Tempest, 1992; Dennis and Gage, 2004; Dennis and Meier, 1997; Hull et al., 1987; Welty et al., 1985; Wu, 1926). Organisms entering through skin or mucous membranes travel via lymphatics to regional lymph nodes, where they multiply. Early infection shows vascular congestion, edema, and minimal inflammation in nodes. Later, nodes contain massive bacteria, vascular damage, hemorrhagic necrosis, and neutrophil infiltration, forming buboes surrounded by serous fluid, often blood-tinged. Multiple nodes may coalesce into a boggy mass, potentially leading to abscess formation and rupture.
Untreated Y. pestis infection can spread to any organ, causing widespread tissue destruction. Common findings in fatal cases include diffuse myocarditis, liver necrosis, spleen and lymph node hemorrhagic necrosis, and renal glomeruli fibrin thrombi (Butler, 1972; Dennis and Meier, 1997; Finegold, 1968). Pneumonitis, pleuritis, and meningitis are less frequent. Abscesses can form in affected organs. Disseminated intravascular coagulation (DIC) leads to microthrombi, thrombocytopenia, necrosis, and bleeding (Butler, 1972). Skin petechiae and ecchymoses are common, as are mucosal and serosal hemorrhages. Late-stage ischemia and gangrene of extremities may occur (Dennis and Meier, 1997; Pollitzer, 1954; Wu, 1926). Primary plague pneumonia from inhalation starts as lobular, progressing to lobar and multilobar consolidation. Alveoli and secretions are bacteria-rich. Secondary pneumonia from hematogenous spread begins as interstitial, becoming diffuse with pulmonary congestion, hemorrhagic necrosis, and neutrophil infiltration (Wu, 1926). Advanced untreated cases show alveoli filled with fluid and massive bacilli.
Clinical Spectrum
Bubonic Plague
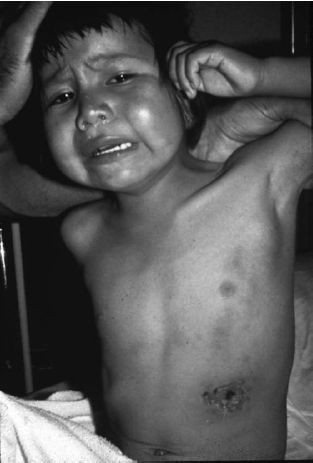
Bubonic plague, the most common form, is characterized by buboes—swollen, tender, inflamed lymph nodes (Butler, 1972). Incubation is typically 2–6 days. Onset is sudden, with chills, rapid fever rise to 100.4°F (38°C) or higher, headache, myalgia, arthralgia, and lethargy. Buboes appear within hours of symptom onset, with increasing swelling, tenderness, and pain in regional lymph nodes near the entry site. Femoral and inguinal nodes are most common, followed by axillary and cervical. About 90% of cases involve a single site, but multiple sites or generalized lymphadenopathy can occur. Patients guard the bubo against palpation and restrict movement. Surrounding tissue often becomes edematous, and overlying skin reddened, warm, tense, and sometimes desquamated. Plague buboes are distinguished by rapid onset, extreme tenderness, edema, toxemia signs, and absence of cellulitis or lymphangitis. Skin inspection may reveal the inoculation site as a papule, pustule, scab, or ulcer (phlyctenule). A large furuncular lesion with eschar may occasionally be present (Figure 2.1). In a study of 40 Vietnamese bubonic plague patients (Butler, 1972):
- Fever (100%; mean 39.4°C)
- Bubo (100%): groin (88%); axilla (15%); cervical (5%); epitrochlear (3%)
- Headache (85%)
- Prostration (75%)
- Chills (40%)
- Anorexia (33%)
- Vomiting (25%)
- Cough (25%)
- Rash (petechiae, purpura, papules) (23%)
- Abdominal pain (18%)
- Chest pain (13%)
Altered brain function (lethargy, confusion, delirium, seizures) was also common. With appropriate antibiotics, uncomplicated plague resolves quickly, with fever and systemic symptoms subsiding in 2–5 days. Buboes may remain enlarged for weeks and sometimes suppurate, requiring drainage. Untreated bubonic plague progresses to severe toxemia, high fever, tachycardia, prostration, agitation, confusion, and potentially convulsions and delirium. Pre-antibiotic era fatality was over 50%; now it is about 5%. Mild bubonic plague, pestis minor, with mild fever and subacute buboes, occurs in South America and elsewhere (Legters et al., 1970), possibly due to immunological tolerance. Serological studies suggest subclinical Y. pestis infections in endemic areas (Ratsitorahina et al., 2000a, 2000b). Differential diagnoses include streptococcal/staphylococcal adenitis, tularemia, cat scratch disease, mycobacterial infection, filarial lymphadenitis, fungal infections, chancroid, sexually transmitted diseases causing lymphadenitis, and strangulated inguinal hernia.
Septicemic Plague
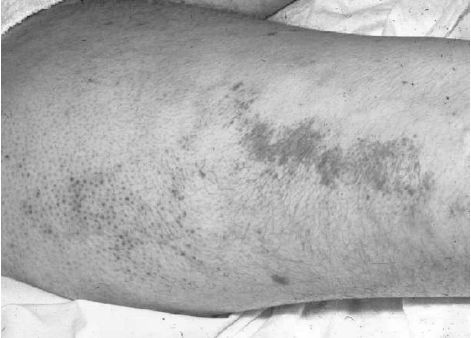
Septicemic plague is a rapidly progressive, overwhelming endotoxemia (Butler et al., 1976; Hull et al., 1987). Primary septic plague occurs without localized infection signs, from direct Y. pestis entry or flea bites. Secondary septic plague arises from bubonic or pneumonic plague when lymphatic or pulmonary defenses are breached, leading to bloodstream multiplication. Bacteremia is common in all plague forms; septicemia is less frequent but life-threatening. Primary septic plague diagnosis is often delayed until blood culture results, as clinical signs are non-specific. Occasionally, bacteria are visible in blood smears, indicating poor prognosis (Butler et al., 1976; Hull et al., 1987; Mann et al., 1984). Gastrointestinal symptoms like nausea, vomiting, diarrhea, and abdominal pain can obscure diagnosis (Hull et al., 1986). Untreated septic plague is fulminant and fatal. Petechiae, ecchymoses, bleeding, and gangrene are manifestations of DIC (Figure 2.2). Refractory hypotension, renal failure, stupor, and shock are preterminal. Acute respiratory distress syndrome can occur and may be confused with hantavirus pulmonary syndrome.
Presenting septicemic plague symptoms in a New Mexico case series (Hull et al., 1987) included:
- Fever (100%; mean 38.5°C, range 35.4–40.4°C)
- Gastrointestinal symptoms (72%)
- Chills (61%)
- Vomiting (50%)
- Nausea (44%)
- Headache (44%)
- Diarrhea (39%)
- Abdominal pain (39%)
Due to late diagnosis, septicemic plague fatality in the US is ≥25% in treated cases and near 100% without antibiotics (Crook and Tempest, 1992; Dennis and Chow, 2004; Hull et al., 1987). Differential diagnoses include other systemic infections, Gram-negative sepsis, meningococcemia, and bacterial endocarditis.
Pneumonic Plague
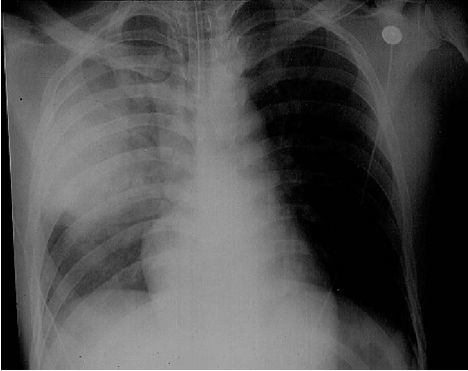
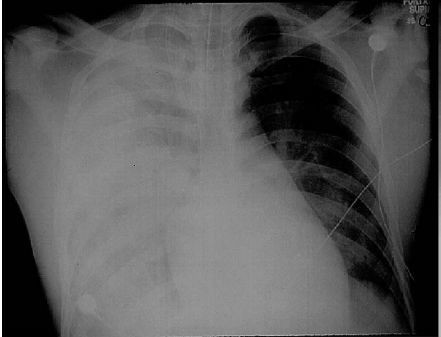
Pneumonic plague is the most rapid and fatal form (Doll, 1994; Laforce et al., 1971; Meyer, 1961; Ratsitorahina et al., 2000a; Tieh et al., 1948; Wu, 1926; Wu et al., 1922; Wynne-Griffith, 1948). Primary pneumonic plague incubation is 3–5 days (range 1–7 days) (Wu, 1926; Wu et al., 1922; K. Alibek, personal communication). Onset is sudden with severe headache, chills, fever, tachycardia, body pains, weakness, dizziness, and chest discomfort. Abdominal pain, nausea, and vomiting may occur. Cough, sputum, chest pain, tachypnea, and dyspnea dominate by day 2, with bloody sputum, respiratory distress, cardiopulmonary insufficiency, and circulatory collapse. Sputum is initially watery or mucoid, frothy, and blood-tinged, becoming frankly bloody. Chest signs may initially be localized, with rapid segmental consolidation progressing to bronchopneumonia (Figure 2.3). Liquefaction necrosis and cavitation may develop, leaving scars.
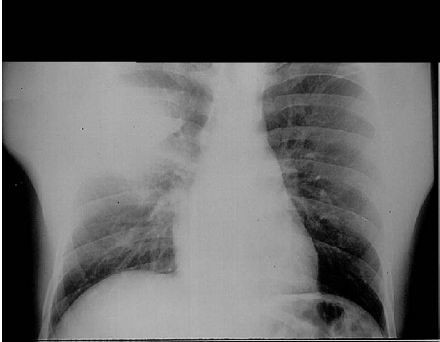
Secondary pneumonic plague often presents as interstitial pneumonitis initially, with scant sputum, progressing rapidly. Chest radiographs show alveolar infiltrates and pleural effusions in many cases (Alsofrom et al., 1981). Advanced cases develop refractory pulmonary edema and sepsis. Person-to-person pneumonic plague transmission has not been recorded in the US since 1924, despite over 50 cases and thousands of potential exposures (Centers for Disease Control, 1984; Centers for Disease Control and Prevention, unpublished data). Differential diagnoses include other bacterial pneumonias, mycoplasma pneumonia, Legionnaire’s disease, staphylococcal/streptococcal pneumonia, tularemia pneumonia, Q fever, severe viral pneumonia, hantavirus pulmonary syndrome, and acute respiratory syndrome from coronavirus infection.
Other Clinical Syndromes
Meningitis is an uncommon plague manifestation. In the US (1947–2001), 17 of 421 plague cases (0.4%) were meningitis, mostly in children (Dennis and Chow, 2003). Meningitis typically occurred late in treated bubonic plague, with high survival rates (Becker et al., 1987; Centers for Disease Control and Prevention, 1997; Mann et al., 1982). Rarely, meningitis occurs without buboes. Symptoms include fever, meningismus, altered mental status, and purulent cerebrospinal fluid with neutrophils; endotoxin may be present (Butler et al., 1976). Pharyngitis can occur from inhalation or ingestion of Y. pestis (Christie et al., 1980), including from contaminated meat. Asymptomatic pharyngeal colonization also occurs (Marshall et al., 1967; Tieh et al., 1948). Rare ocular plague cases exist, from conjunctival inoculation or systemic spread (Poland, 1989). Primary ocular and pharyngeal plague could result from intentional Y. pestis release.
Pediatric Plague
Pediatric plague is clinically similar to adults, but children are more likely to develop systemic spread (septicemia, pneumonia, meningitis) and have higher fatality rates (Burkle, 1973; Mann et al., 1982). A US review of 183 childhood cases (1947–2001) (Dennis and Chow, 2003) found 91% bubonic plague, with 32% developing complications (sepsis, pneumonia, meningitis). Mortality was highest in primary sepsis/pneumonia (39%), less in bubonic plague with secondary spread (30%), and lowest in uncomplicated bubonic plague (9%). Children are more likely to develop dissemination complications (32% vs. 27% in adults), especially meningitis, and have higher fatality rates (17% vs. 14%). Diagnosis delay is common in children; in one series, plague was considered in <10% at first encounter and ~40% at admission (Mann et al., 1982). Presenting symptoms included:
- Fever (95%)
- Lethargy, malaise, anorexia (40%)
- Vomiting (50%)
- Chills (29%)
- Headache (29%)
- Abdominal discomfort (26%)
- Diarrhea (8%)
Plague in Pregnancy
Plague in pregnant women can cause fetal infection, stillbirth, abortion, and perinatal infection (Mann and Moskowitz, 1977; Welty et al., 1985).
Diagnosis
Laboratory Diagnosis
Laboratory Response Capabilities
Plague laboratory tests are highly reliable when performed by trained personnel, previously limited to few reference labs. The US established the Laboratory Response Network (LRN) in 1999 to enhance standardized diagnostic testing for plague and select agents in response to bioterrorism concerns (Centers for Disease Control and Prevention, 2000; Morse et al., 2003). This network links state and local public health labs with advanced labs at CDC, NIH, FDA, DoD, FBI, and USDA. Member labs are sentinel (Level A) or reference (Levels B-D), with increasing safety and technical capabilities. Sentinel (Level A) labs, including hospital labs, use BSL-2 safety procedures for initial Y. pestis identification or rule-out using staining, culture, and biochemical tests. Suspicious samples are sent to Level B and C labs (public health labs with BSL-2/3) for advanced rapid diagnostics, confirmation, and strain characterization. Level B/C labs also perform antimicrobial susceptibility testing, and Level C labs conduct molecular subtyping (Henchal et al., 2001; Morse et al., 2003) for epidemiology and forensics.
Collection and Processing of Specimens
Clinical specimens (blood, bubo aspirates, sputum, tracheal washes, lesion swabs, CSF) should be collected promptly for microbiology (Henchal et al., 2001; Miller, 2001), chest radiographs taken, and antibiotics started pending diagnosis confirmation. Inoculate specimens into media (brain-heart infusion broth, sheep blood agar, chocolate agar, MacConkey agar). Collect blood cultures before antibiotics. Blood culture counts range from <10 to 4 × 10^7 CFU/mL. Bubo aspirates yield small serosanguinous fluid; inject 1–2 mL saline and aspirate for diagnosis. Prepare smears for Gram and polychromatic stains (Wayson or Giemsa). Direct fluorescent antibody testing is a rapid presumptive test in specialized labs. Collect acute-phase serum for Y. pestis antibody testing, followed by convalescent-phase serum in 3–4 weeks. Plague diagnosis can be confirmed by a four-fold or greater antibody titer change to Y. pestis antigen using passive hemagglutination, or a single titer ≥1:128 in a compatible illness without plague vaccination. Antibodies may be detectable as early as 5 days after onset, with most seroconverting in 1–2 weeks, some later, and a few not seroconverting (1977; Centers for Disease Control and Prevention, unpublished data). Early antibiotics may delay seroconversion. Positive titers decline over months to years. Enzyme-linked immunosorbent assays (ELISA) for IgM and IgG antibodies help detect early infection and differentiate from vaccine-induced antibodies. Presumptive Y. pestis identification can be made by PCR or antigen-capture ELISA (Henchal et al., 2001; Loiez et al., 2003; Radnedge et al., 2001; Rahalison et al., 2000). A rapid immunogold dipstick assay shows promise for rapid bedside diagnosis even in resource-limited settings (Chanteau et al., 2003). Clinical lab protocols for plague and select agent diagnosis are available for bioterrorism events (American Society of Microbiology, Biological Weapons Resources Center, 2003; Centers for Disease Control and Prevention, Bioweapons Laboratory Issues, 2001; Henchal et al., 2001). Nonspecific lab findings include elevated liver/heart enzymes, thrombocytopenia, and leukocytosis (10,000–25,000/mm3) with neutrophil predominance. Leukemoid reactions (>50,000/mm3 WBC) can occur (Butler et al., 1974; Welty et al., 1985).
For fatal cases, collect autopsy tissues (lymph nodes, liver, spleen, lungs, bone marrow) for culture, fluorescent antibody testing, and histology with immunohistochemical staining (Guarner et al., 2002). Cary Blair medium can be used to transport tissues for Y. pestis isolation.
Recognizing a Plague Outbreak Resulting from Intentional Release
Rapid identification of intentional plague release is critical to prevent mortality and secondary transmission. This requires expertise in clinical, laboratory, and public health services. Frontline healthcare professionals and local health departments are key for early detection and reporting (Ashford et al., 2003). Table 2.1 outlines expected features of an aerosol exposure outbreak.
Table 2.1.
Diagnosis of Plague following Release of an Aerosol of Y. pestis
| Epidemiology and Symptoms | Sudden outbreak of geographically linked persons with fever, cough, shortness of breath, hemoptysis, and chest pain; point source exposure pattern, incubation period 1–7 days, peaking 3–5 days postexposure; gastrointestinal symptoms common (e.g., nausea, vomiting, abdominal pain, and diarrhea); rapidly developing toxemia; fulminant and fatal course common. |
|---|---|
| Clinical Signs | Tachypnea, dyspnea, and cyanosis; pneumonic consolidation on chest examination; septic shock and organ failure; disseminated intravascular coagulation, petechiae, ecchymoses; occasional cases of plague pharyngitis and cervical adenitis, conjunctivitis, possible. |
| Laboratory Studies | Sputum, throat swab, tracheal-bronchial washes for Gram, and Wayson or Giemsa staining; culture at 28°C and 37°C on suitable media (e.g., sheep’s blood agar, McConkey’s, chocolate agar, BHI broth); if suspicion high, send above to LRN reference laboratory for DFA, advanced rapid testing; standard blood culture as per institution protocol; if suspicion high, culture subset at 28°C; acute-phase serum specimen to be held at 4°C until plague ruled out; antimicrobial susceptibility testing of Y. pestis isolates; subtyping studies to characterize isolates, e.g., PFGE, PCR, MLVA; virulence testing; chest radiographs. |
| Pathology | Lobular exudation, bacillary aggregation, hemorrhagic necrosis of pulmonary parenchyma; tissues processed for direct detection (DFA, IHC) and isolation of Y. pestis |
Detection of Y. pestis in the Environment
Y. pestis culture is the standard confirmation method, but is slow and insensitive. Rapid tests are needed for early warning, environmental contamination detection, and epidemiological/forensic investigations. Gene amplification systems using PCR to detect Y. pestis DNA (Pst, Cafl, YopM, Pla targets) have been developed (Henchal et al., 2001; Radnedge et al., 2001; Rahalison et al., 2000). Fluorogenic 5′ nuclease chemistry and real-time PCR (e.g., TaqManR) allow rapid detection. Instruments like Lightcycler™, RAPID™, or SmartCycler enable identification in 20–40 minutes after nucleic acid purification (Henchal et al., 2001). “Sniffing devices” for aerosolized pathogens are in development and testing in the US. BioWatch, a PCR-based system, monitors air in major cities for Y. pestis and other select agents. Other systems include IBADS™ (immunoassay) and BIDS™ (light addressable potentiometric device) (Henchal et al., 2001). These technologies, initially for military use, are experimentally used to monitor civilian events and sensitive sites. In case of suspected aerosol release, environmental sampling using PCR, immunochromatographic assay, and culture can identify the agent and contamination extent. Only culture determines agent viability and allows characterization.
Environmental release concerns may extend to rodent and animal infections, posing human risks from animal- or flea-borne plague. Field teams would collect and process animals and fleas for Y. pestis detection using rapid methods and culture. Rodent and flea control measures would be implemented (Gage, 1998; Gratz, 1999). Cat surveillance may be needed as cats are susceptible and can transmit plague, including respiratory infection, to humans (Gage et al., 2000).
Medical Management of Plague Patients
Antimicrobial Treatment of Acute Illness in Naturally Occurring Plague
Untreated plague fatality exceeds 50% for bubonic plague and is near 100% for septicemic or pneumonic plague. US plague mortality in the last 50 years is ~15% (Centers for Disease Control and Prevention, 1997; Craven et al., 1993; Dennis and Campbell, 2004), primarily due to delayed treatment or misdiagnosis (Centers for Disease Control and Prevention, 1997; Crook and Tempest, 1992). Rapid diagnosis and effective antibiotic treatment are essential, ideally within 24 hours of pneumonic symptom onset (Butler, 1994; Butler and Dennis, 2004). Y. pestis strains are generally susceptible to recommended antibiotics: streptomycin, gentamicin, doxycycline, chloramphenicol, and trimethoprim-sulfamethoxazole, and fluoroquinolones (ciprofloxacin, ofloxacin, trovafloxacin) (Frean et al., 1996; Smith et al., 1995; Wong et al., 2000). Ceftriaxone also shows in vitro activity, but clinical experience is limited. Studies in baboons showed doxycycline or ciprofloxacin comparable to streptomycin or tetracycline for bubonic plague, and gentamicin or streptomycin highly effective for pneumonic plague (Romanov et al., 2001a, 2001b). Rifampin, ampicillin, aztreonam, ceftazidime, cefotetan, and cefazolin are less effective for pneumonic plague in mice compared to streptomycin and gentamicin (Byrne et al., 1998).
Streptomycin was the traditional drug of choice (Butler and Dennis, 2004), but is no longer US-manufactured and has limited availability. Gentamicin, readily available and intravenously administrable, is increasingly used in the US. Gentamicin is reported to be effective (Crook and Tempest, 1992; Welty et al., 1985). A retrospective analysis of 50 gentamicin-treated plague patients in New Mexico since 1970 suggests gentamicin (or gentamicin/doxycycline) is as effective as streptomycin (Boulanger et al. 2004). Tetracyclines or chloramphenicol are alternatives. Doxycycline, due to easy administration, absorption, and serum levels, is preferred. Doxycycline treatment should begin with a loading dose (IV or oral). Chloramphenicol is indicated for meningitis, pleuritis, endophthalmitis, or myocarditis, used alone or with aminoglycosides. Trimethoprim-sulfamethoxazole is effective for bubonic plague but less favored as a first-line option due to potentially delayed response. Penicillins, cephalosporins, and macrolides are not recommended. Antibiotic treatment should last 7-10 days, or at least 3 days after afebrile status and clinical recovery (Butler and Dennis, 2004; Dennis, 2001; Inglesby et al., 2000). IV antibiotics can be switched to oral as indicated, usually by day 4 or 5. Improvement is typically seen in 2-3 days, although fever may persist. Antimicrobial-resistant Y. pestis strains are rare and have not been associated with treatment failures, except for the multidrug-resistant strain in Madagascar (Galimand et al., 1997). Table 2.2 provides treatment guidelines for non-bioterrorism settings.
Table 2.2.
Plague Treatment Guidelines in Usual (Nonbioterrorism) Circumstances
| Drug | Dosage | Route of administration |
|---|---|---|
| Streptomycina | ||
| Adults | 1 g q 12 hr | IM |
| Children | 15 mg/kg q 12 hra | IM |
| Gentamicinb | ||
| Adults | 1–1.5 mg/kg q 8 hrc | IV or IM |
| Children | 2.0–2.5 mg/kg q 8 hrc | IV or IM |
| Infants/neonates | 2.5 mg/kg q 8 hrc | IV or IM |
| Tetracyclined | ||
| Adults | 0.5 g q 6 hr | PO |
| Children >8 yr | 6.25–12.5 mg/kg q 6 hr | PO |
| Doxycyclined | ||
| Adults | 100 mg q 12 hr | IV or PO |
| Children >8 yr and >45 kg | 100 mg q 12 hr | IV or PO |
| Children >8 yr and | 2.2 mg/kg q 12 hr | IV or PO |
| Chloramphenicold | ||
| Adults | 12.5 mg/kg q 6 hre | IV or PO |
| Children > 1 yr | 12.5 mg/kg q 6 hre | IV or PO |
Common complications of delayed treatment include DIC and ARDS, requiring intensive care. Investigational sepsis treatments include recombinant-activated protein C (Dellinger, 2003; Wheeler and Gordon, 1999). Ciprofloxacin and sympathetic blockade have been used to treat plague sepsis and gangrene (Kuberski et al., 2003). Abscessed nodes can cause recurrent fever.
Postexposure Prophylaxis
Postexposure antibiotic prophylaxis is recommended for those with unprotected respiratory exposure to pneumonic plague patients (Centers for Disease Control and Prevention, 1996; Inglesby et al., 2000). Treatment is indicated within 7 days of exposure for 7 days. Tetracycline, doxycycline, sulfonamides, and chloramphenicol are recommended (Centers for Disease Control and Prevention, 1996; Poland and Dennis, 1999), with tetracycline and doxycycline FDA-approved for prophylaxis. Fluoroquinolones may also be effective (Russell et al., 1996) and are advocated for bioterrorism response (Inglesby et al., 2000). Short prophylaxis courses may be advised for household members of bubonic plague patients. Prophylaxis is rarely needed for residents in plague areas. Respiratory plague patients are generally noncontagious after 48 hours of antibiotics (Anonymous, 2000; Inglesby et al. 2000).
Treatment of Cases and Case Contacts in a Bioterrorism Event
A plague aerosol attack necessitates rapid pneumonic plague case identification and treatment. The Department of Homeland Security’s National Pharmaceutical Stockpile (NPS) contains antimicrobials and materials for plague management. The Johns Hopkins Working Group provides guidelines for bioterrorism-related plague treatment and prophylaxis using NPS resources (Inglesby et al., 2000), considering both “contained casualty” (individualized care, parenteral antibiotics) and “mass casualty” (oral antibiotics for large numbers). In contained situations, streptomycin or gentamicin are preferred, with parenteral doxycycline, ciprofloxacin, and chloramphenicol as alternatives. Oral therapy is substituted upon improvement. In mass casualty events, oral doxycycline and ciprofloxacin are preferred, with chloramphenicol as an alternative (Table 2.3). Postexposure prophylaxis regimens are the same as mass casualty treatment (oral doxycycline or ciprofloxacin) but for 7 days (Table 2.4).
Table 2.3.
Plague Treatment Guidelines Using National Pharmaceutical Stockpile Components
| Initial therapy | Duration |
|---|---|
| ADULTS | Gentamicin 5 mg/kg IM or IV once daily (or in 3 divided doses)a–e OR |
| Parenteral | Doxycycline 100 mg IV twice daily OR |
| Oral | Doxycycline 100 mg PO twice daily |
| CHILDREN | Gentamicin 7.5 mg/kg IM or IV once daily (or, in 3 divided doses)a–e OR |
| Parenteral | Doxycyclinef >45 kg: 100 mg IV twice daily ≤45 kg: 2.2 mg/kg IV twice daily OR Doxycyclinef |
| Oral | >45 kg: 100 mg PO twice daily ≤45 kg: 2.2 mg/kg PO twice daily |
| PREGNANCY | Same as for nonpregnant adultsg,h |
| IMMUNOCOMPROMISED | Same as for nonimmunocompromised adults and children |
Table 2.4.
Plague Prophylaxis Guidelines Using National Pharmaceutical Stockpile Componentsa
| Therapy | Duration |
|---|---|
| ADULTS | Doxycycline 100 mg PO twice daily OR Ciprofloxacin 500 mg PO twice dailyb,c |
| CHILDREN | Doxycyclined >45 kg: 100 mg PO twice daily ≤45 kg: 2.2 mg/kg PO twice daily OR Ciprofloxacin 20 mg/kg PO twice dailybce |
| PREGNANCY | Same as for nonpregnant adultsf |
| IMMUNOCOMPROMISED | Same as for nonimmunosuppressed adults and children |
Treatment criteria include fever ≥38.5°C or new cough in potentially exposed individuals in affected communities, and tachypnea in young children. Persons with fever or cough during prophylaxis should be evaluated for plague and potential resistance. Postexposure prophylaxis is recommended within 7 days for: aerosol exposure, household members of respiratory plague cases, healthcare workers, emergency responders, transporters of suspected cases, close contacts of respiratory plague cases, release site investigators, and public health personnel involved in investigations. Pre-exposure prophylaxis and respiratory masks may be considered for high-risk groups.
Infection Control
Hospital Infection Control
Current consensus is that pneumonic plague transmission is via respiratory droplets (>5 μm), not airborne transmission (<5 μm). Respiratory droplets are generated by coughing, sneezing, talking, and procedures like suctioning and bronchoscopy, transmitted over short distances (<2 meters). Respiratory Droplet Precautions and Standard Precautions (eye protection, gloves, mask, gowns) are essential within 2 meters of contagious patients (Grow and Rubinson, 2003). Patients should be moved from isolation only when necessary, masked, and avoiding contact. CDC website provides hospital infection control guidelines (Centers for Disease Control and Prevention, Hospital Infections Program, 2003).
The Role of Isolation and Quarantine
Isolation, separating infected individuals during communicability, is recommended for confirmed, probable, or suspect pneumonic plague cases for the first 48 hours of antibiotics and until improvement or rule-out. Alternative isolation facilities may be needed if hospital capacity is exceeded, with security, utilities, medical care, and communication. Asymptomatic exposed individuals should receive prophylaxis and monitoring. Isolation may not be needed for asymptomatic contacts on prophylaxis, but they should avoid close contacts and activities that could spread infection for the first 48 hours of prophylaxis.
Quarantine, restricting movement of well individuals exposed to communicable diseases, is used to prevent transmission. Legal basis for quarantine is both state and federal. The Model State Emergency Health Powers Act (Johns Hopkins Center for Civilian Biodefense Strategies, 2003; Mair et al. 2002) addresses state powers for quarantine during biological attacks, including facility closures and hospital takeover. Quarantine measures for intentional Y. pestis release may include public gathering suspension, public place closures, travel restrictions, and cordon sanitaire (perimeter control) (World Health Organization, 1983). Travel restrictions and cordon sanitaire should be implemented cautiously due to potential infringement on rights, fear, and civil disobedience.
Prevention
Prevention and Control of Naturally Occurring Plague
The WHO recommends a four-phase plague prevention and control system (Gage, 1999; World Health Organization, 1980, 1983): 1) Case recognition and intervention; 2) Epidemiological/epizootiological investigation and emergency control; 3) Predictive surveillance and preventive control; 4) Management. Endemic areas should have continuous human and animal surveillance, investigations, and control actions. Environmental remediation measures during outbreaks include flea control, rodent control, and sanitation. Flea control should precede or accompany rodent killing to minimize human flea exposure (Gage, 1998; Gratz, 1999a). Outbreak control measures include: source identification, geographic limit definition, active surveillance, lab confirmation and pneumonic case isolation, rapid treatment, flea and rodent control, and traveler screening from epidemic areas (Fritz et al., 1996).
Plague Vaccine
A killed, whole-cell plague vaccine has limited availability and utility. A comparable vaccine is produced by CSL Ltd. (Australia). The initial adult course is two 0.5-mL doses at 1-4 week intervals, followed by 6-monthly boosters (Titball et al., 2004). Use is recommended for high-risk groups: lab workers, biologists in enzootic areas, and some military personnel. Efficacy against flea-borne exposure is thought to be present, but protection against respiratory exposure is limited (Centers for Disease Control and Prevention, 1996; Titball et al., 2004). Live attenuated vaccines, used in the past by the Soviet Union, India, and former French colonies, offer single-dose, early but incomplete protection against both routes, requiring annual revaccination. They have high adverse reaction rates. Live vaccines are not commercially available; Russia uses a live vaccine (EV strain) for annual vaccination of high-risk individuals (K. Alibek, personal communication). Research focuses on improved vaccines protective against airborne exposure (Titball et al., 2004; Williamson, 2001). Recombinant subunit vaccines expressing F1 and V antigens are promising (Titball and Williamson, 2003), showing protection against aerosol exposure in animals. Inhalable aerosol vaccine formulations are being developed (Eyles et al., 2000). Bioterrorism concerns have increased interest in effective plague vaccines.
Research Directions
Plague emergency preparedness drives research in early agent detection (bioluminescent protein detectors, improved sniffing devices), rapid diagnostic markers (hand-held assays), rapid organism characterization (genomic and proteomic advances), improved treatments (bactericidal/permeability increasing proteins), shock countermeasures (recombinant-activated protein C), and recombinant inhalant/DNA vaccines and postexposure immunoprotective agents (monoclonal antibodies) (Beamer, 2002; Casadevall, 2002; Chanteau et al., 2003; Dellinger, 2003; Eyles et al., 2000; McDonnell and Askari, 1996; Parkhill et al., 2001; Rider et al. 2003; Wheeler and Gordon, 1999).