Introduction
In 1983, groundbreaking research highlighted that the pain hypersensitivity associated with tissue injury or inflammation wasn’t solely a peripheral issue but significantly involved the central nervous system (CNS) [255]. This concept, although foreshadowed a century earlier by W Allen Sturge MD in 1883, who proposed a central nervous system “commotion” in angina pectoris, remained largely unexplored for decades. My research demonstrated that peripheral injury could trigger a lasting increase in the excitability of spinal cord neurons, fundamentally altering the somatosensory system’s sensitivity [255]. This central amplification manifested as reduced pain thresholds (allodynia), heightened and prolonged responses to noxious stimuli (hyperalgesia), and expanded receptive fields causing pain from previously non-painful areas (secondary hyperalgesia) [255–256; 273; 51; 268].
Recently, I reviewed the circumstances surrounding the discovery of activity-dependent synaptic plasticity in the spinal cord, which underlies post-injury pain hypersensitivity [259]. This phenomenon became known as “central sensitization” [272]. I have also explored the cellular and molecular mechanisms driving this neuronal plasticity [147]. In this review, I aim to delve into the clinical implications of central sensitization. What insights has it provided into the nature and mechanisms of patient pain? Crucially, what are the Central Sensitization Implications For The Diagnosis And Treatment Of Pain? To address these questions, we must first define central sensitization, understand how it revolutionized our understanding of pain, and examine the substantial evidence from experimental pain studies in human volunteers.
What is Central Sensitization?
Before the discovery of central sensitization, the CNS’s role in pain processing was viewed as a passive relay, transmitting information about noxious stimuli—onset, duration, intensity, location, and quality—much like a telephone wire. The “pain pathway,” a specific anatomical network in the spinal cord, brain stem, thalamus, and cortex, was believed to link sensory input from high-threshold primary afferents to cortical areas responsible for pain awareness. Melzack and Wall’s gate control theory in 1965 introduced the concept of spinal cord modulation by inhibitory controls [163], and by the early 1980s, significant progress was made in identifying these inhibitory circuits [18]. Discoveries like enkephalins and endorphins [109; 98], diffuse noxious inhibitory controls [150], transcutaneous nerve stimulation [224], and acupuncture [25] shifted focus towards endogenous inhibitory mechanisms rather than factors that might increase excitation and cause pain hypersensitivity. Peripheral sensitization, discovered in the 1970s [178], was a notable exception. Iggo [112; 28] and Perl [33; 177; 20] identified nociceptors—specialized high-threshold sensory neurons responding exclusively to noxious stimuli [265], a term Sherrington coined based on flexion reflex studies. Perl and others demonstrated that nociceptor peripheral terminals could become “sensitized” after injury, reducing their threshold, mainly to heat, within the injury site—the primary hyperalgesia zone [178; 146; 138; 41; 23]. While crucial in inflammatory pain hypersensitivity [22], peripheral sensitization couldn’t explain dynamic tactile allodynia, temporal summation of pain, or secondary hyperalgesia. A different explanation was needed, which turned out to be increased synaptic function within the CNS triggered by nociceptive inputs [257; 237; 268].
By the early 1980s, the idea that synapses could undergo use-dependent plasticity, increasing their strength, was gaining traction. Lloyd described post-tetanic potentiation of IA synaptic input to motor neurons in 1949 [155], spreading to other motor neuron synapses [21]. Mendell and Wall’s 1965 discovery of windup in dorsal horn neurons showed that repeated low-frequency C-fiber stimulation caused progressively increased action potential firing [164]. A major breakthrough was Bliss and Lomo’s 1973 description of long-term potentiation (LTP) in the hippocampus, where brief high-frequency input caused persistent synaptic efficacy increase, sparking extensive research into synaptic plasticity’s molecular mechanisms. LTP was first recorded in the spinal cord in 1993 [182], representing a component of central sensitization [113; 122; 114]. In 1976, Kandel and colleagues described gill withdrawal reflex sensitization in Aplysia, linked to facilitated synapses between sensory and motor neurons [29]. This was interpreted as memory and learning, not pain hypersensitivity, though these phenomena overlap [274; 122].
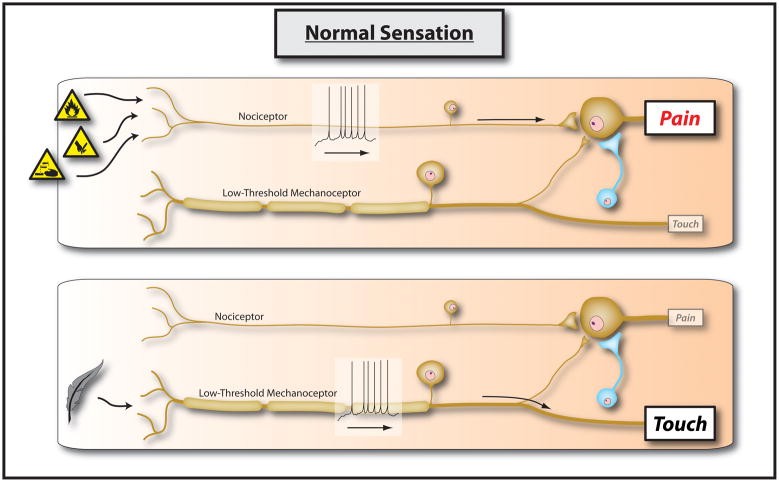
My 1983 and subsequent preclinical studies at University College London revealed that brief (10–20 seconds), low-frequency (1–10Hz) bursts of action potentials into the CNS, from electrical stimulation or natural nociceptor activation, increased synaptic efficacy in dorsal horn nociceptive neurons. This effect lasted for tens of minutes after the conditioning stimulus [255; 244; 256; 267; 50; 245; 273; 51; 272; 263; 230; 264]. Unlike windup, which is increasing output during stimulus trains (homosynaptic potentiation), central sensitization is facilitation after conditioning stimuli, sustaining itself or needing only minimal nociceptor input. It involves input from one set of nociceptor fibers (conditioning input) amplifying responses to other non-stimulated non-nociceptor or nociceptor fibers (test input)—heterosynaptic potentiation [231]. Hippocampal LTP is typically homosynaptic, with efficacy changes at activated synapses. While some central sensitization aspects are homosynaptic [190], clinically relevant attributes are largely heterosynaptic [147]. The neurobiological basis is that most central circuits have “iceberg” receptive fields. Most synaptic input is subthreshold [262–263], subliminal due to weak input or inhibition-restrained membrane excitability. Increased synaptic strength via presynaptic excitatory transmitter release, postsynaptic response [264; 271; 231; 129; 247; 130; 133; 154; 46; 100; 151–152; 227], reduced inhibition [208; 168; 12; 103; 180; 165; 226], or increased membrane excitability can recruit subthreshold inputs to suprathreshold action potentials, profoundly altering function [270]. Recent understanding includes contributions from microglia, astrocytes, gap junctions, membrane excitability, and gene transcription to central sensitization maintenance [234; 189; 43; 48; 88; 104; 205; 44; 47; 186]. Figures 1 and 2 illustrate normal sensory processing and changes from central sensitization.
Figure 1.
Figure 1: Normal sensation. The somatosensory system is structured so that specialized primary sensory neurons encoding low-intensity stimuli activate pathways leading to innocuous sensations only. High-intensity stimuli activating nociceptors activate pathways leading to pain. These parallel pathways do not functionally overlap, maintained by strong synaptic inputs and inhibitory neurons focusing activity within dedicated circuits.
Figure 2.
Figure 2: Central sensitization. Induction of central sensitization in somatosensory pathways leads to increased synaptic efficacy and reduced inhibition. This central amplification enhances the pain response to noxious stimuli in amplitude, duration, and spatial extent. Strengthening normally ineffective synapses allows subliminal inputs, such as low-threshold sensory inputs, to activate the pain circuit. The two parallel sensory pathways converge.
A key implication of these early studies was that pain might not always reflect a peripheral noxious stimulus. We learn to interpret pain as indicating peripheral damage, crucial for its protective function. Central sensitization introduces a dimension where the CNS can alter pain, increasing its intensity, duration, and spread beyond what peripheral stimuli dictate, reflecting CNS circuit states rather than specific peripheral stimuli. Central sensitization “centralizes” pain, uncoupling the direct stimulus-response relationship of nociceptive pain. Nociceptive pain is the perception of noxious stimuli; without such stimuli, there is no nociceptive pain. However, central sensitization reveals that noxious stimuli, while sufficient, are not necessary for pain. Increased gain in CNS “pain pathway” neurons can cause activation by low-threshold, innocuous inputs. Pain, in these cases, becomes an illusory perception, identical to that from real noxious stimuli but occurring without them. This pain is real but not nociceptive; it is induced pain hypersensitivity, closely resembling many clinical conditions. This immediately raised the question: is central sensitization involved in clinical pain hypersensitivity?
Initially, these ideas were poorly received, especially by physicians who attributed pain without pathology to malingering, drug seeking, or psychiatric issues. The notion of central pain amplification as a “real” neurobiological phenomenon contributing to clinical pain conditions was met with skepticism. Clinicians often used vague diagnoses like psychosomatic or somatoform disorder for unexplained pain. Now, 30 years later, data from human volunteer and patient studies allow us to assess whether central sensitization—defined operationally as CNS neural signaling amplification causing pain hypersensitivity—is real and its role in inflammatory, neuropathic, and dysfunctional pain disorders [258; 53].
Central Sensitization in Human Volunteers
The first clear evidence of central sensitization in humans came from LaMotte and colleagues’ psychophysical study on secondary cutaneous hyperalgesia induced by intradermal capsaicin (TRPV1 receptor activator). They observed intense localized pain at the injection site, followed by three hyperalgesia zones: a small, short-lived (1–2 hours) heat hyperalgesia zone near the injection site, an intermediate, longer-lasting (several hours) dynamic tactile allodynia zone spreading beyond heat hyperalgesia, and a largest pinprick hyperalgesia zone, far from the injection site, lasting up to 24 hours [145]. They demonstrated that secondary mechanical hyperalgesia required CNS sensory inflow, as local anesthesia before capsaicin blocked it. Furthermore, pain sensitivity crossing a tight circulation-preventing skin band indicated it wasn’t due to local capsaicin spread or peripheral inflammatory mediators. La Motte and Torebjork’s 1992 study further confirmed activity-dependent central sensitization’s role in tactile allodynia and secondary hyperalgesia [233]. Using intradermal capsaicin to induce 2-hour tactile allodynia, nerve block experiments showed C fibers mediated capsaicin and heat pain, but low-threshold myelinated fibers transmitted mechanical allodynia to the CNS. Elegantly, they found that electrical intraneural stimulation of single Aβ mechanoreceptive fibers, eliciting non-painful tactile sensation before capsaicin, began to produce pain if the fibers’ receptive field fell within secondary mechanical hyperalgesia zones. Lidocaine anesthesia of the stimulated fiber’s cutaneous innervation area didn’t reverse the pain, indicating non-peripheral origin. They concluded that pain from stroking skin around a painful intradermal capsaicin injection “is due to reversible changes in the central processing of mechanoreceptive input from myelinated fibres which normally evoke non-painful tactile sensations”.
Koltzenburg and Torebjork’s study using mustard oil (TRPA1 activator) and differential nerve blocks also confirmed that brush-evoked mechanical allodynia was mediated by low-threshold Aβ fibers normally encoding non-painful tactile sensations [140]. Unlike capsaicin, mustard oil-evoked tactile allodynia needed ongoing low-level C-nociceptor input, suggesting different sensory fibers have varied central actions—some short, others long-lasting. Subsequent studies showed varying durations of tactile allodynia after capsaicin and mustard oil [139], whose significance was not immediately clear as irritants acted on different TRP receptors.
Max and colleagues demonstrated that central sensitization could spread pain sensitivity across peripheral nerve territories, a neurological indicator of central rather than peripheral nervous system disease, using intradermal capsaicin with radial or ulnar nerve blocks to delineate nerve territories [192]. A study comparing skin hyperaemia from burn injury found that skin blood flow changes disappeared by the time secondary mechanical hyperalgesia peaked, and they weren’t temporally or spatially correlated, supporting non-peripheral mechanisms for secondary hyperalgesia [198]. Remarkably, intradermal capsaicin induced contralateral hyperalgesia and allodynia, delayed and less extensive than ipsilateral secondary hyperalgesia, but present in most subjects [206], a “tertiary hyperalgesia” form unlikely to be peripheral. Our pain sensitivity is thus determined by CNS neuron excitability.
Central amplification of Aδ nociceptor fiber test input after C-fiber conditioning input contributes to pinprick/punctate secondary hyperalgesia, as shown using intradermal capsaicin [279], highlighting distinct afferent signals for conditioning (C-fibers) and eliciting allodynia (Aβ) or hyperalgesia (Aδ)—heterosynaptic facilitation. Similarly, pinprick hyperalgesia from intradermal capsaicin was mediated by capsaicin-insensitive afferents, showing different test and conditioning inputs [87]. Secondary hyperalgesia from intradermal capsaicin was restricted to mechanical stimuli, with no correlation between capsaicin-evoked pain magnitude and punctate or tactile secondary hyperalgesia extent [237]. Temporal summation to pinprick in the capsaicin injection zone (homosynaptic facilitation/windup model) was mechanistically independent of secondary hyperalgesia development. While stimulus-response gain in the secondary zone increased, windup gain remained unchanged, though actual pain was enhanced [158]. Repeated intradermal capsaicin injections produced diminishing pain, likely due to desensitization, while allodynia and punctate hyperalgesia continued to increase [254]. Recent studies using high-frequency stimulation to mimic LTP-inducing conditions found that while changes occur at the conditioned site (homotopic), pain hypersensitivity develops at adjacent non-stimulated heterotopic sites (reduced threshold, tactile pain, exaggerated pinprick response) [136; 240]. Both groups concluded heterosynaptic facilitation predominates in this central sensitization model, as with low-frequency conditioning mimicking natural nociceptor firing. Heterosynaptic changes appear to be a major feature of central sensitization presentation.
Besides subjective pain measures, central sensitization’s effects can be detected with objective biomarkers, including long-term changes in nociceptive withdrawal reflexes [24] and increased cortical event-related potential amplitudes [240]. Magnetic source imaging reveals increased somatosensory cortex neuron excitability from low-threshold Aβ stimulation within capsaicin-induced secondary hyperalgesia zones [17], while magnetoencephalography detects cerebral processing pattern changes [159], and fMRI detects cortical BOLD signal changes during secondary hyperalgesia [16]. Another MRI study found brainstem changes specific to central sensitization, alongside primary somatosensory cortex changes related to pain intensity [153].
While most studies examine skin conditioning stimulus effects on skin pain sensitivity, experimental muscle pain from hypertonic saline injections causes lasting thermal sensitivity changes in referred pain areas [203]. Sustained nociceptive stimulation of myofascial trigger points induces widespread central sensitization [273; 275]. Preclinical models show muscle and joint conditioning afferents have longer-lasting central sensitization effects than skin afferents [244]. Cutaneous capsaicin increases myofascial trigger point pressure sensitivity in segmentally related muscles [211]. Visceral conditioning stimuli, like esophageal acid exposure, induce central sensitization, causing viscerovisceral (upper esophageal pain hypersensitivity) and viscerosomatic hypersensitivity (chest wall allodynia) [193], captured by esophageal evoked potentials [194] and associated with increased temporal summation [196]. A recent esophageal central sensitization model using acid and capsaicin infusions also showed rectal thermal and mechanical pain hypersensitivity after esophageal stimulation [27], highlighting widespread gastrointestinal effects. These changes may relate to widespread clinical pain syndromes [95].
Experimental central sensitization in humans is increasingly used to test centrally acting drug efficacy. Drugs effective in preclinical models, like NMDA receptor antagonists [271], can be tested in Phase 1b human proof-of-principle studies [212]. Ketamine inhibits central temporal summation [8] and secondary mechanical hyperalgesia [142] evoked by repetitive nociceptive electrical stimulation, as well as primary and secondary hyperalgesia after experimental burn injury [116], visceral conditioning inputs [253; 251], and topical [6] or intradermal [204] capsaicin, but not A delta-mediated nociceptive pain [181]. Ketamine’s action on experimental pain is detectable by fMRI [210]. IV dextromethorphan shows similar activity [115]. These data strongly support NMDA receptor involvement in acute activity-dependent central sensitization [147]. However, trials also indicate a narrow therapeutic index between reducing central sensitization and causing psychotomimetic side effects. Gabapentanoids are another extensively studied drug class in human experimental central sensitization models. Oral gabapentin, at chronic neuropathic pain doses, reduced tactile allodynia and mechanical secondary hyperalgesia from intradermal capsaicin [92]. Even single gabapentin doses had antihyperalgesic effects on capsaicin-induced secondary hyperalgesia and reduced fMRI signatures of central sensitization [110]. Gabapentin reduced cutaneous-evoked central sensitization but not muscle pain [201]. Pregabalin’s efficacy in human experimental central sensitization was studied in electrical stimuli [49] and intradermal capsaicin models [246]. Both double-blind studies showed pregabalin efficacy against tactile allodynia and secondary hyperalgesia, suggesting central sensitization reduction is a major mechanism for gabapentin and pregabalin [238]. Many other centrally acting analgesics effective in patients, such as duloxetine, milnacipran, and lamotrigine [118; 170; 15], reduce central sensitization preclinically but haven’t been tested in humans for this effect. NK1 receptor antagonists [252, 49] and COX-2 inhibitors [35; 250; 49] have failed to show efficacy in human activity-dependent central sensitization studies. However, COX-2 inhibitors are effective if central sensitization is triggered by peripheral inflammation [225], as preclinical models predict [189].
While gender is noted for nociceptive pain sensitivity differences, a study on heat and capsaicin-induced secondary hyperalgesia found no gender difference [119]. However, recent data indicate heritability of pain sensitivity, including secondary hyperalgesia and brush-evoked allodynia, with about 50% genetic contribution to pain variance [172]. Genetic polymorphisms influencing secondary hyperalgesia susceptibility are not fully explored, though some candidates are emerging from experimental central sensitization studies [228]. This area needs significant research.
From human volunteer experimental pain studies, we conclude: Central sensitization is robust, readily induced by diverse nociceptor activation methods (electrical stimulation, capsaicin, mustard oil, acid, heat burn, UV burn, hypertonic saline). This activity-dependent plasticity manifests immediately and persists for hours after the conditioning stimulus, eventually reverting to baseline, indicating reversibility. It can be induced by conditioning skin, muscle, or viscera, typically presenting as dynamic tactile allodynia and punctate hyperalgesia, but also enhanced pressure and sometimes thermal sensitivity, spreading from the conditioning site to adjacent and remote areas. While homosynaptic (homotopic) aspects exist, its major manifestation is heterosynaptic (heterotopic), making equating central sensitization with LTP-like phenomena in cortex associated with long-term memory inaccurate due to reversibility. Because central sensitization is inducible in most subjects, detectable by subjective and objective measures, and sensitive to pharmacological interventions, it’s a useful tool for assessing drug activity on centrally driven pain hypersensitivity.
Human volunteer studies demonstrate that use-dependent central facilitation in nociceptive pathways increases pain sensitivity and may contribute to clinical pain syndromes. Experimental studies are limited to non-injurious conditioning inputs, studying only activity-dependent pain hypersensitivity components, not transcription-dependent and structural changes after inflammation or nerve injury, which may have different mechanisms, time courses, and presentations [269; 171; 121; 160; 261; 189; 123; 229; 97; 53; 242]. Limited severe human experimental injury experience indicates central sensitization also contributes to late hyperalgesia in these models [58; 176].
Central Sensitization and the Clinical Pain Phenotype
What clinical phenotype features might be caused or contributed to by central sensitization? Human experimental studies suggest that dynamic tactile allodynia, secondary punctate/pressure hyperalgesia, temporal summation, and sensory aftereffects indicate potential central sensitization involvement. Any sensory experience exceeding expected amplitude, duration, and spatial extent from a defined peripheral input under normal circumstances could reflect central amplification from increased excitation or reduced inhibition. These include reduced thresholds, exaggerated noxious stimulus responses, post-stimulus pain, and sensitivity spread to normal tissue. However, direct sensory inflow measurement is impossible, and peripheral changes like peripheral sensitization can amplify sensation. Therefore, pain hypersensitivity alone isn’t definitive for central sensitization diagnosis. Peripheral input often triggers central sensitization, so pain sensitivity reduction by targeting a peripheral trigger with local anesthetic doesn’t exclude central amplification but might indicate peripheral input’s role in maintaining it [140]. Nonetheless, some patient symptom features are more indicative of central than peripheral contributions: pain mediated by low-threshold Aβ fibers (nerve block or electrical stimulation determined), pain sensitivity spread to areas without demonstrable pathology, aftersensations, enhanced temporal summation, and pain maintenance by normally non-painful low-frequency stimuli. To assess central sensitization presentation in patients, detailed phenotyping of different cohorts is needed to capture sensitivity changes—what, where, when [93; 188; 86; 9; 11; 197; 55]. Ideally, this should be combined with objective central activity measures like fMRI to establish clear diagnostic criteria for central sensitization in patients. Diagnostic criteria for central sensitization would provide insights into pathophysiological mechanisms and guide treatment strategies. If pain primarily results from abnormal nociceptor activity, as in primary erythromelalgia patients [74], optimal therapy differs from patients with tactile allodynia and secondary hyperalgesia maintained by central sensitization due to spinal cord synaptic efficacy changes. This is the rationale for a mechanism-based pain diagnosis and treatment approach [266; 258]. Trial treatment response, like to NMDA receptor antagonist ketamine, can itself be diagnostic for central sensitization.
To which clinical syndromes does central sensitization contribute?
Despite caveats about definitive diagnostic criteria, numerous studies have identified central sensitization as contributing to patient pain phenotypes across various syndromes.
Rheumatoid Arthritis (RA)
RA patients, with prototypic inflammatory joint disease, have extra-articular tenderness correlated with joint disease extent [141], but whether this is peripheral or central sensitization is unstudied. Juvenile chronic arthritis studies reported enhanced noxious stimuli sensitivity at joints and remote areas in patients with and without active disease, suggesting active disease may induce autonomous central sensitization [[107](#R107]].
Osteoarthritis (OA)
OA, a degenerative joint disease with cartilage destruction and bone alteration, is a common chronic pain cause, especially in the elderly. Pain severity doesn’t always correlate with joint damage or inflammation, suggesting a central pain component [26]. Supporting this, OA patients show enhanced pain duration and secondary hyperalgesia from intramuscular hypertonic saline injection compared to controls [13]. High preoperative pain and low pain thresholds increase persistent pain risk after total knee replacement for OA, interpreted as central sensitization [157]. Another study of 62 patients showed central neural origin pain (widespread reduced pressure pain thresholds) negatively impacted knee functional capacity [117]. OA patients have lower pain thresholds and punctate hyperalgesia in referred pain areas, associated with greater brainstem activation on fMRI, a potential biomarker for central changes [99]. Duloxetine, a centrally acting amine uptake inhibitor reducing central sensitization in preclinical models [124; 15], significantly reduced pain more than placebo in a knee OA pain RCT of 231 patients [45], indicating central sensitization-targeting drugs are effective in this population. A phenotyping study of 48 painful knee OA patients and 24 age-matched controls showed patients had reduced pressure pain thresholds at joints and remote areas, and increased temporal summation. Sensitization degree correlated with pain but not radiological findings, concluding central sensitization is a key contributor to knee OA pain [7]. OA pain, despite peripheral pathology, appears to have a significant central component, warranting further study for extent, mechanism, and therapeutic implications.
Temporomandibular Disorders (TMD)
Unlike OA, TMD pathophysiology is less understood. TMD is associated with increased generalized pain sensitivity after orofacial muscle isometric contraction [166]. Widespread bilateral mechanical [78] and thermal [175] pain sensitivity is reported in women with myofascial TMD compared to age-matched controls, suggesting widespread central sensitization. More frequent trigger points in these patients elicit greater referred pain than in controls [77].
In facial pain, mechanical allodynia is prominent in periradicular inflammation (periradicular periodontitis), with reduced thresholds in contralateral non-inflamed teeth, reflecting central sensitization [132]. After third molar extraction, central sensitization evidence persisted for at least a week (enhanced repetitive intraoral pinprick and electrical stimulation response, aftersensations, extraoral hyperalgesia) [126].
Fibromyalgia (FM)
Early suggestions of generalized central sensitization in fibromyalgia patients came from psychophysical studies identifying widespread thermal and mechanical pain threshold reductions, and greater cerebral laser-evoked potentials [90], soon replicated [156]. An early small ketamine study showed an NMDA-dependent component in fibromyalgia, suggesting tender points may represent secondary hyperalgesia from central sensitization [209]. Arendt-Nielson and colleagues found fibromyalgia patients had lower pressure thresholds, increased temporal summation to muscle stimulation, and intramuscular hypertonic saline injections provoked longer-lasting, more widespread pain. Relatedly, they found ketamine attenuated referred pain, temporal summation, muscular hyperalgesia, and muscle pain in fibromyalgia patients [96]. In 2001, Staud and Price began fibromyalgia studies, first showing temporal summation and aftersensations of pain from repetitive cutaneous thermal stimuli and muscle mechanical stimuli [221]. They then found temporal summation occurred at lower forces and frequencies in fibromyalgia patients than controls, with greater and prolonged painful aftersensations [215]. Enhanced experimental pain in fibromyalgia patients contributed to clinical pain variance [220]. They showed experimentally induced pain maintenance in fibromyalgia patients requires less frequent stimulation than in controls, concluding heightened sensitivity to low-frequency inputs contributes to persistent pain [218]. Later, they showed temporal summation of pain and its maintenance were widespread, elicitable equally from hands or feet, suggesting generalized central sensitization across the neuraxis [219]. An fMRI study showed stimulus and frequency-dependent activation in several brain regions in fibromyalgia patients and controls (ipsilateral and contralateral thalamus, medial thalamus, S1, bilateral S2, mid- and posterior insula, rostral and mid-anterior cingulate cortex). However, stimulus temperatures for equivalent brain activity were lower in fibromyalgia patients, suggesting enhanced neural mechanisms aren’t solely cortical [216]. Staud and Price’s group explored peripheral sensitization’s contribution to enhanced thermal pain temporal summation in fibromyalgia patients, concluding it doesn’t contribute based on thermal thresholds [214]. Recent local anesthetic injection studies suggest peripheral muscle input is important for maintaining central sensitization in FM patients [217]. Fibromyalgia may have both peripheral and central contributions, varying by patient. Muscle afferents appear potent in preclinical [244] and human experimental studies [275] for inducing central sensitization.
Quantitative sensory testing in 85 fibromyalgia patients and 40 matched controls showed altered heat and cold thresholds, reduced pain tolerance, and reduced nociceptive reflex threshold—a central excitability measure [65]. The latter was distinct enough from controls to suggest diagnostic potential for central sensitization, identifying patients benefiting from centrally acting drugs. Other studies confirmed increased generalized sensitivity in FM patients to pressure and thermal stimuli [179; 94; 173] and skin and muscle electrical stimulation, with enhanced cortical evoked potentials [66]. Overall data supports a major role for central sensitization in fibromyalgia symptoms, and centrally acting treatments like pregabalin or duloxetine may be effective by reducing central sensitization.
Miscellaneous Musculoskeletal Disorders
Chronic neck pain from whiplash is associated with lowered pain thresholds in uninjured tissue [57; 222]. Local anesthetic injection into myofascial trigger points in these patients immediately increases range of motion and pressure pain thresholds, reflecting dynamic central sensitization maintenance by afferent triggers [85]. Shoulder impingement syndrome patients also show widespread muscle sensitivity and increased trigger points [105]. Unilateral epicondylalgia (tennis elbow) patients exhibit widespread (bilateral) mechanical pain hypersensitivity, suggesting central sensitization induced by a peripheral trigger [75]. Similar generalized deep tissue hyperalgesia occurs in chronic radiating low back pain patients with intervertebral disc herniation [173]. These data suggest diverse musculoskeletal disorders involve pain sensitivity spread to deep uninjured tissue, maintained by low-level peripheral inputs.
Headache
Initial indications of central sensitization in headaches came from spontaneous tension-type headache studies showing increased pericranial muscle tenderness in patients even without headache, compared to controls. During headache, muscle tenderness increased and temporal region thermal pain threshold decreased, but hand thresholds remained normal, suggesting segmental central sensitization contributes to frequent tension-type headache pain [120]. Bernstein and colleagues then observed cutaneous allodynia in 79% of migraine attack patients, sometimes beyond referred pain areas [36–37]. This has been replicated in several studies [161; 135; 207; 52]. Cephalic and extracephalic allodynia are well-described, and spontaneous body pain and allodynia preceding migraine attacks have been reported [56]. Laser-evoked cutaneous pain thresholds are reduced during migraine attacks, and cortical evoked potentials are increased [62]. No heat pain threshold changes are found in chronic tension-type headache, but pericranial tenderness [63; 80] and neck and shoulder muscle hyperalgesia exist [81]. Muscle nociceptive input may induce central sensitization in tension-type headache [79], similar to fibromyalgia. Cluster headache patients show reduced nociceptive flexion reflex threshold on the symptomatic side [191]. A population study on primary headaches in 523 patients found pain hypersensitivity evidence in tension-type pain, greater in chronic or frequent headaches, implying central sensitization may contribute to headache chronification [30], supported by epidemiological data [31]. A longitudinal prospective study of 100 individuals found normal thresholds before headache development, decreasing in those developing chronic tension-type headache, suggesting pain hypersensitivity is a consequence of frequent tension-type headaches, not a predictor [32], indicating central sensitization’s role in tension-type headache chronification. A study of chronic migraine and chronic tension-type headache patients found reduced thresholds for pressure, pinprick, blink, and nociceptive flexion reflex, and higher windup ratios in both cohorts [83], possibly reflecting a common central sensitization role in different headache types’ chronification.
Neuropathic pain
Early evidence for central sensitization in neuropathic pain came from Campbell and colleagues, showing ischemic conduction block of large myelinated fibers specifically reduced dynamic tactile allodynia [42], soon replicated [140]. Carpal tunnel syndrome phenotyping studies revealed enhanced bilateral sensitivity and extraterritorial symptom spread in unilateral or single nerve entrapment, supporting central sensitization contribution [61; 76; 82; 278]. Ketamine reduces established peripheral neuropathic pain [125] and chronic phantom limb pain [73], indicating ongoing activity- and NMDA receptor-dependent synaptic plasticity may maintain neuropathic pain. Tricyclic antidepressants, dual uptake inhibitors, and calcium channel alpha(2)-delta ligands—centrally acting drugs normalizing enhanced neural activity—are first-line neuropathic pain treatments [72], reinforcing the central pain component’s importance and its suitability as a treatment target.
Complex Regional Pain Syndrome (CRPS)
Chronic CRPS1 prominently features tactile hyperesthesia and pressure hyperalgesia [241], registered as enhanced S1 activation by neuromagnetometer [243]. Acute CRPS1 patients also have thermal hyperalgesia, which may have a peripheral component on the diseased limb side due to aseptic inflammation, but contralateral hypersensitivity without inflammation points to CNS involvement [108]. A small randomized placebo-controlled trial showed intravenous ketamine reduced CRPS pain [200].
Post-surgical pain
Post-surgical pain is heterogeneous, including acute postoperative pain and persistent pain from multiple causes, including surgical-induced neuropathic pain [131; 1]. Acute incisional pain is associated with ketamine-sensitive secondary punctate hyperalgesia [223], without thermal sensitivity spread [143], indicating central sensitization induction. Pre-emptive treatment targeting central sensitization versus postoperative treatment for acute postoperative pain or chronic pain transition is controversial [260; 68; 128; 60; 149; 70–71; 102; 4–5; 54; 236]. Design, conduct, and interpretation issues make resolution difficult [167; 134]. While pre-emptive analgesia literature is vast, my interpretation is a small signal for pre- vs. postoperative analgesic treatment in some settings, but likely not generally clinically relevant. Ensuring full analgesia upon general anesthesia recovery or adequate regional anesthesia during surgery, maintained until surgical healing is advanced, is crucial [19; 277; 14]. Postoperative pain control can include central sensitization-acting drugs like ketamine [184], pregabalin [162; 34], gabapentin [202], and duloxetine [106], showing some efficacy in limited trials, but more RCTs are needed to assess their utility in acute postoperative pain or chronic pain risk reduction [59].
Visceral Pain Hypersensitivity Syndromes
Pain hypersensitivity characterizes gastrointestinal disorders like irritable bowel syndrome, non-cardiac chest pain, and chronic pancreatitis, all with central sensitization components. Most IBS patients have rectal and somatic hypersensitivity [249]. Repetitive sigmoid stimulation in IBS patients induces rectal hyperalgesia and viscerosomatic referral [169]. Local rectal anesthesia reduces rectal and somatic pain in IBS patients, suggesting visceral hyperalgesia and secondary cutaneous hyperalgesia in IBS result from central sensitization dynamically maintained by GIT input. Non-cardiac chest pain patients have esophageal hypersensitivity [195], reduced tolerance to repeated distension, increased referred pain size, and greater secondary hyperalgesia propensity after lower esophageal acid infusion [69], reflecting central sensitization consequences. Chronic pancreatitis is associated with generalized deep pressure hyperalgesia [39; 174], and patients show greater degree and spatial extent secondary hyperalgesia from repetitive experimental stimulation, suggesting enhanced central sensitization [67], reduced by thorascopic splanchnic denervation [38], indicating pancreatic visceral input maintains central sensitization.
In the urological tract, pain hypersensitivity features interstitial cystitis, chronic prostatitis, endometriosis, and vulvodynia, conditions with poorly understood pathophysiology and etiology. Central sensitization is hypothesized to contribute [137], but data is limited. Chronic prostatitis men have heightened perineal pain sensitivity [276; 239], while vulvodynia women have enhanced post-capsaicin allodynia and secondary hyperalgesia compared to controls [84].
Co-morbidity of pain conditions characterized by pain hypersensitivity
Pain is nociceptive from noxious stimuli, inflammatory from tissue injury/immune activation, and neuropathic from nervous system lesions. But what about pain conditions without noxious stimuli, inflammation, or nervous system damage? Several common syndromes present with pain hypersensitivity without clear etiology, considered “unexplained,” potentially reflecting primary nervous system dysfunction rather than peripheral pathology. These include fibromyalgia, tension-type headache, temporomandibular joint disease, and irritable bowel syndrome, all possibly having central sensitization contributions. If heightened CNS sensitivity or central sensitization propensity is common in these syndromes, comorbidity would be expected. Enhanced central sensitization capacity may be the primary defect in some syndromes.
A study of almost 4,000 twins on comorbidity of chronic fatigue, low back pain, irritable bowel syndrome, chronic tension-type headache, temporomandibular joint disease, major depression, panic attacks, and post-traumatic stress disorder found associations far exceeding chance, concluding shared etiology [199]. Another large epidemiological twin study on 44,000 individuals on chronic widespread pain comorbidity found co-occurrence with chronic fatigue, joint pain, depressive symptoms, and irritable bowel syndrome, suggesting genetic factors in chronic widespread pain and comorbidity associations [127]. Another study of 2299 subjects on four unexplained syndromes—chronic widespread pain, chronic orofacial pain, irritable bowel, and chronic fatigue—again found greater multiple syndrome occurrence than expected by chance [2]. These epidemiological findings strongly suggest a common mechanistic basis for these diverse conditions, potentially hereditary.
Smaller studies show fibromyalgia comorbidity with migraine in females but not males [111], primary headache [64], chronic fatigue symptom [89], systemic lupus erythematosus [213], irritable bowel syndrome [144], rheumatoid arthritis [183], premenstrual syndrome [3], chronic urticaria [235], and cervical myofascial pain syndrome [40]. Comorbidity is also shown for back pain and temporomandibular disorders [248], migraine and temporomandibular disorders [91], irritable bowel syndrome and functional dyspepsia, fibromyalgia and chronic pelvic pain [185], and migraine, irritable bowel syndrome, chronic fatigue, and fibromyalgia [232]. Overlap also exists between urological disorders like chronic pelvic pain, interstitial cystitis, painful bladder syndrome, chronic prostatitis, and vulvodynia with fibromyalgia, chronic fatigue, temporomandibular disorders, and irritable bowel syndrome [187], specifically between vulvodynia, fibromyalgia, and irritable bowel syndrome [10].
These diverse epidemiological studies overwhelmingly conclude that chronic pain hypersensitivity without inflammation or nerve damage results in phenotypically different syndromes based on affected tissues/organs. However, similar sensitivity changes may reflect common central sensitization contributions, explaining unexpectedly high comorbidity rates of seemingly different syndromes. To test for central sensitization syndromes, clear diagnostic criteria and biomarkers are needed. If this hypothesis is correct, treatment strategies normalizing CNS hyperexcitability may have shared efficacy across different central sensitization syndrome manifestations.
Diagnostic Criteria and Biomarkers for Central Sensitization
Developing diagnostic criteria for central sensitization is crucial for improving pain management. Currently, diagnosis relies on clinical features and quantitative sensory testing (QST).
Clinical Features suggestive of Central Sensitization:
- Disproportionate Pain: Pain intensity and duration exceeding what is expected from the apparent peripheral injury or pathology.
- Diffuse Pain Distribution: Pain spreading beyond the expected anatomical distribution of the initial injury.
- Allodynia: Pain response to normally non-painful stimuli (e.g., light touch).
- Hyperalgesia: Increased pain response to painful stimuli.
- Temporal Summation: Increased pain with repeated stimulation.
- Aftersensations: Pain persisting after the removal of the stimulus.
- Pain Referral: Pain perceived in areas distant from the site of actual tissue irritation or injury.
- Variability of Pain: Fluctuations in pain intensity and location over time.
- Comorbidities: Presence of other central sensitivity syndromes like fatigue, IBS, or headache.
Quantitative Sensory Testing (QST) findings in Central Sensitization:
- Reduced pressure pain thresholds: Indicating widespread hyperalgesia to pressure.
- Enhanced temporal summation: Demonstrating increased central excitability.
- Allodynia to light touch: Assessing abnormal pain processing of innocuous stimuli.
- Increased pain intensity ratings: In response to suprathreshold stimuli.
- Altered thermal thresholds: In some cases, indicating broader sensory dysregulation.
Potential Biomarkers for Central Sensitization:
- Functional MRI (fMRI): Studies show altered brain activity patterns in central sensitization conditions. Specific regions like the brainstem and somatosensory cortex show changes related to pain processing and central amplification [153, 16]. fMRI could potentially serve as an objective measure of central sensitization, although further research is needed to standardize protocols and validate findings for clinical use.
- Magnetoencephalography (MEG): MEG can detect changes in cerebral processing patterns and cortical excitability in central sensitization [[159](#R159], 17]. MEG offers high temporal resolution and could complement fMRI in characterizing central sensitization.
- Electrophysiological Measures: Increased cortical event-related potential amplitudes and changes in nociceptive withdrawal reflexes can serve as objective biomarkers of central sensitization [[240](#R240], 24. These measures reflect altered central nervous system responses to stimuli.
- Genetic Markers: Research is emerging on genetic polymorphisms that may contribute to differential susceptibility to central sensitization [[228](#R228], 172. Identifying these genetic markers could help identify individuals at higher risk of developing central sensitization and related pain conditions.
- Neurochemical Markers: Further research into neurochemical changes in the CNS associated with central sensitization (e.g., neurotransmitter levels, inflammatory mediators in the cerebrospinal fluid) may reveal potential biomarkers.
Standardized diagnostic criteria, combining clinical features, QST findings, and potentially biomarkers, are essential for accurate identification of central sensitization in patients. This will facilitate targeted treatment approaches and improve patient outcomes.
Treatment Strategies Targeting Central Sensitization
Understanding central sensitization as a key mechanism in chronic pain has significant implications for treatment. Therapeutic strategies should aim to normalize hyperexcitability in the CNS and address the various mechanisms contributing to central sensitization.
Pharmacological Treatments:
- NMDA Receptor Antagonists: Ketamine and dextromethorphan have shown efficacy in reducing central sensitization in experimental and clinical settings [142, 115]. Ketamine, in particular, has been used to treat neuropathic pain, CRPS, and fibromyalgia, likely through its action on NMDA receptors, which are crucial in synaptic plasticity and central sensitization [200, 209. However, side effects, especially psychotomimetic effects, limit their widespread use.
- Gabapentanoids: Gabapentin and pregabalin are widely used for neuropathic pain and fibromyalgia. They reduce central sensitization by modulating calcium channels and reducing excitatory neurotransmitter release [238]. Studies have shown their effectiveness in reducing tactile allodynia and secondary hyperalgesia in experimental central sensitization and clinical pain conditions [92, 246].
- Serotonin-Norepinephrine Reuptake Inhibitors (SNRIs): Duloxetine and milnacipran, SNRIs, are effective in treating fibromyalgia, chronic musculoskeletal pain, and neuropathic pain. They are believed to reduce central sensitization by enhancing descending inhibitory pathways and modulating neurotransmitter levels in the CNS [124, 15].
- Tricyclic Antidepressants (TCAs): TCAs like amitriptyline have been used for chronic pain conditions, including neuropathic pain and headache. They have complex mechanisms, including neurotransmitter modulation and potential effects on central sensitization pathways.
- Opioids: While opioids are effective for acute pain, their role in chronic pain, especially conditions involving central sensitization, is more complex. Opioids can have limited effectiveness and potential risks in chronic non-cancer pain. In some cases, they may even exacerbate central sensitization (opioid-induced hyperalgesia).
- Cannabinoids: Cannabinoids are being explored for chronic pain management, including conditions with central sensitization. They interact with the endocannabinoid system, which plays a role in pain modulation and central nervous system excitability. However, more research is needed to define their efficacy and safety in central sensitization syndromes.
Non-Pharmacological Treatments:
- Physical Therapy and Exercise: Exercise and physical therapy can improve function and reduce pain in chronic pain conditions. Physical activity may modulate central sensitization through various mechanisms, including endorphin release and improved descending inhibition.
- Psychological Therapies: Cognitive Behavioral Therapy (CBT), Acceptance and Commitment Therapy (ACT), and other psychological therapies can help patients cope with chronic pain. These therapies can address psychological factors contributing to pain perception and central sensitization, such as stress, anxiety, and maladaptive coping strategies.
- Mindfulness and Meditation: Mindfulness-based interventions can modulate pain processing and reduce central sensitization by altering brain activity patterns and enhancing descending inhibitory pathways.
- Transcutaneous Electrical Nerve Stimulation (TENS): TENS can activate inhibitory circuits in the spinal cord and reduce pain. It may have some effect on modulating central sensitization mechanisms.
- Acupuncture: Acupuncture may activate descending inhibitory pathways and modulate central nervous system excitability. It has been used in chronic pain management and may have some benefits in conditions involving central sensitization.
- Manual Therapy: Techniques like massage and spinal manipulation may reduce pain and improve function in musculoskeletal pain conditions. Manual therapy may have some effects on modulating central sensitization, although the mechanisms are not fully understood.
- Neuromodulation Techniques: Spinal cord stimulation and other neuromodulation techniques are used for chronic pain management. These techniques can directly modulate nervous system activity and may reduce central sensitization in certain conditions.
Multimodal and Personalized Treatment Approaches:
Optimal management of conditions involving central sensitization often requires a multimodal approach, combining pharmacological and non-pharmacological treatments. Personalized treatment plans, tailored to the individual patient’s specific pain mechanisms, comorbidities, and psychosocial factors, are essential. Identifying patients with predominant central sensitization features is crucial for selecting appropriate treatments targeting central mechanisms.
Future Directions in Treatment:
- Targeting Specific Molecular Mechanisms: Ongoing research into the molecular mechanisms of central sensitization may lead to the development of more targeted pharmacological treatments. For example, drugs specifically targeting specific ion channels, neurotransmitter receptors, or inflammatory mediators involved in central sensitization.
- Biomarker-Guided Treatment: Using biomarkers to identify patients with central sensitization and to monitor treatment response could improve treatment precision and outcomes.
- Combination Therapies: Combining different pharmacological and non-pharmacological treatments that target different aspects of central sensitization may lead to synergistic effects and improved pain relief.
- Preventive Strategies: Identifying risk factors for developing central sensitization and implementing preventive strategies may reduce the burden of chronic pain conditions.
Conclusions
Clinical pain is significantly influenced by the excitability of central nociceptive circuits, not just peripheral pathology. Activity-dependent synaptic function increases in these circuits, triggered and maintained by nociceptor inputs, alter the pain system’s sensitivity. This leads to normally innocuous inputs activating the system, and exaggerated, prolonged, and widespread responses to noxious inputs. These sensory changes are central sensitization manifestations. Extensive experimental and clinical research over two decades has confirmed its importance in pain hypersensitivity in many patients. While cellular and molecular mechanisms are increasingly understood [148], much remains to be discovered, especially regarding genetic and environmental risk factors, triggers, maintenance, and persistence. Nevertheless, identifying central sensitization’s contribution to many “unexplained” clinical pain conditions offers both mechanistic explanations and therapeutic targets. Recognizing the central sensitization implications for the diagnosis and treatment of pain is vital for advancing pain management and improving the lives of millions suffering from chronic pain.
Acknowledgments
Supported by research funds from the NIH. I thank all my colleagues whose work over the past 25 years has contributed to central sensitization study, especially Alban Latremoliere for manuscript review and Christian von Hehn for figure creation.
Footnotes
There is no conflict of interest.
Publisher’s Disclaimer: This is a PDF file of an unedited manuscript accepted for publication. Provided as a customer service, this early manuscript version will undergo copyediting, typesetting, and proof review before final citable publication. Note that production process errors may affect content, and all journal legal disclaimers apply.
References
References are in the original article and are not copied here to maintain focus on the rewritten content as per instructions. The original reference list should be appended to the final markdown document.