1. Understanding Cerebral Folate Deficiency Syndrome (CFDS)
Cerebral Folate Deficiency Syndrome (CFDS) encompasses a spectrum of neuropsychiatric and developmental disorders characterized by diminished folate levels within the cerebrospinal fluid (CSF), despite normal folate levels outside the central nervous system. The clinical presentation of CFDS is remarkably varied, influenced significantly by the timing of folate deficiency – whether it occurs in the womb or postnatally. A primary driver of CFDS is the presence of serum folate receptor-alpha (FRα) autoantibodies, which impede the crucial transport of folate across the choroid plexus into the brain. However, CFDS can also arise from mitochondrial disorders, inherited metabolic errors, and loss-of-function mutations in the FOLR1 gene, which encodes FRα. Early and accurate Cfd Diagnosis is paramount for effective intervention and improved patient outcomes. This article delves into FRα autoimmunity, its diverse age-related clinical syndromes, the essential diagnostic criteria for cfd diagnosis, and available treatments, including preventative strategies for at-risk populations.
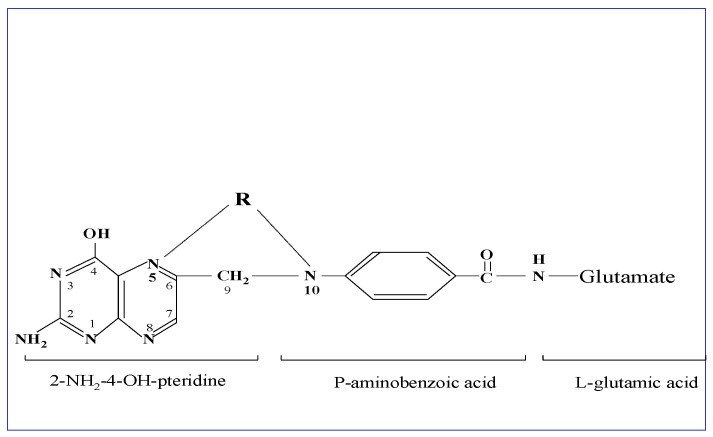
Figure 1: Structural Formula of Folic Acid. Depicting the core components: a 2-amino-4-hydroxypteridine molecule linked to p-aminobenzoylmono-glutamate via methylene, and a variable one-carbon group (R) at different oxidation states. Tetrahydrofolate, the active form, features reduction at positions 5,6,7,8 of the pteridine ring.
2. The Crucial Role of Folate and its Transport in Neurological Health
Folic acid, or folate (vitamin B9), is not a single entity but a group of interconvertible forms vital for fertility, embryonic and fetal development, and postnatal neurological function. Folates participate in numerous metabolic processes and epigenetic mechanisms, including gene expression regulation. Vitamins B2, B6, and B12 are essential cofactors for enzymes involved in folate interconversion pathways, such as homocysteine degradation, the remethylation and methionine cycles, the transsulfuration cycle, and the synthesis of purines and thymidylate. Adequate levels of these cofactors and proper enzyme function are essential for the various roles of folate, including cell proliferation, homocysteine metabolism, neurotransmitter synthesis, and epigenetic regulation.
Figure 2: Folate Metabolism Pathways in the Brain. Illustrating 5-methyl-tetrahydrofolate (5-methyl THF) transport across the choroid plexus via FRα, its role in methionine and remethylation cycles, and the connection to neurotransmitter synthesis and antioxidant pathways. Key enzymes and metabolites are highlighted, emphasizing the central role of folate in brain function.
Folate absorption predominantly occurs in the jejunum, facilitated by the proton-coupled folate transporter (PCFT). A smaller fraction is absorbed via the reduced folate carrier (RFC1). Within intestinal cells, folic acid is converted to tetrahydrofolate (THF) and then to N5-methyl-tetrahydrofolate (MTHF), the primary circulating form. MTHF is transported across the blood-brain barrier at the choroid plexus by FRα and PCFT, a process critical for maintaining brain folate levels. This transport is essential for neuronal function and overall neurological health. Similar FRα-mediated transport is also crucial at the placenta, gonads, and thyroid gland, especially during fetal development and early infancy.
3. Hereditary Folate Malabsorption and the Concept of Cerebral Folate Deficiency
The understanding of folate deficiency dates back to the 1930s when Lucy Wills identified a treatable anemia linked to yeast extract, later found to be due to folic acid. Intrauterine folate deficiency is linked to neural tube defects and increased risk of developmental issues, including autism. Severe systemic folate deficiency can lead to macrocytic anemia, immune dysfunction, and neurological disorders. Hereditary folate malabsorption, caused by genetic defects in PCFT, exemplifies the systemic consequences of folate deficiency. This condition impairs both intestinal folate absorption and folate transport to the brain, resulting in a severe clinical picture with systemic and neurological manifestations. Early diagnosis and folinic acid treatment can significantly mitigate neurological complications in hereditary folate malabsorption.
In 2004, the term Cerebral Folate Deficiency (CFD) was introduced to define a distinct entity: neuropsychiatric disorders associated with low CSF MTHF levels despite normal systemic folate status. This definition underscores that serum folate levels are not reflective of brain folate status, making CSF MTHF measurement essential for accurate cfd diagnosis.
Several mechanisms can lead to CFD, affecting folate availability in the brain (Table 1). These include impaired folate transport across the blood-brain barrier, reduced folate storage or release within the brain, increased folate utilization or catabolism in the nervous system, and metabolic conditions disrupting folate metabolism specifically in the brain.
Table 1: Mechanisms Underlying Cerebral Folate Deficiency (CFD). Outlining five categories of mechanisms that can lead to CFD, ranging from transport deficits and storage issues to increased utilization and metabolic disturbances within the nervous system.
Table 2 provides a broader overview, listing conditions associated with both systemic folate depletion and CFD, highlighting the diverse origins of folate-related neurological issues.
Table 2: Systemic Folate Depletion and Cerebral Folate Deficiency (CFD) Conditions. Categorizing conditions leading to systemic folate deficiency versus those specifically causing CFD, emphasizing the isolated folate loss in the CNS in CFDS.
Infantile-onset CFDS, the first described CFD syndrome, shares neurological similarities with hereditary folate malabsorption but lacks the systemic hematological, intestinal, and immunological abnormalities seen in the latter. Infantile CFDS presents with neurological symptoms arising from isolated brain folate deficiency. Typically, onset occurs between 4-6 months of age, with initial signs including sleep disturbances and agitation, progressing to psychomotor retardation, hypotonia, ataxia, pyramidal signs, and potentially dyskinesias or seizures. Untreated individuals may develop visual and hearing loss. Unlike hereditary folate malabsorption, brain scans in infantile CFDS do not show basal ganglia calcifications. MRI scans in some untreated cases reveal delayed myelination and cerebral/cerebellar atrophy. Routine laboratory tests are usually normal, emphasizing the need for specific CSF folate measurement for cfd diagnosis.
4. Systemic Folate Deficiency: Causes and Contributing Factors
Systemic folate deficiency is a widespread issue, often stemming from malnutrition globally. Poor dietary intake, especially in conditions like alcoholism and psychiatric disorders with eating disturbances, can lead to significant folate depletion. Malabsorption disorders, such as celiac disease, Crohn’s disease, and chronic intestinal infections, can also impair folate absorption in the jejunum.
Certain medications, particularly antifolate drugs used in cancer chemotherapy (methotrexate, 5-fluorouracil, pemetrexed), directly interfere with folate metabolism and utilization. Other drugs, including isoniazid, sulfonamides, anticonvulsants (valproic acid, phenytoin, carbamazepine), and carbidopa, can also disrupt folate metabolism or cellular uptake, contributing to systemic deficiency. Congenital folate malabsorption, a rare genetic condition due to PCFT gene mutations, is another cause of systemic folate deficiency, as discussed previously.
Inborn errors of metabolism, such as methylene-THF reductase (MTHFR) deficiency, can also impact systemic folate levels. MTHFR deficiency, particularly severe forms, can lead to hyperhomocysteinemia and other complications. Dihydrofolate reductase (DHFR) deficiency and glutamate formiminotransferase deficiency are other rare inherited metabolic conditions that can result in systemic folate deficiency with varying clinical manifestations. Recently, methylenetetrahydrofolate dehydrogenase-1 (MTHFD1) deficiency has been identified as a cause of megaloblastic anemia and variable hyperhomocysteinemia, further expanding the spectrum of inherited folate metabolism disorders.
5. Cerebral Folate Deficiency (CFD): Unraveling the Etiology
In infantile-onset CFDS, the most common underlying cause is now recognized as serum autoantibodies against folate receptor-alpha (FRαAb). These autoantibodies impair MTHF binding and transport across the choroid plexus, leading to reduced folate levels in the CSF and brain. Two types of FRαAbs are recognized: blocking antibodies, which directly interfere with folate binding to FRα, and binding antibodies, which bind to FRα at a different site but still disrupt its function, potentially through complement-mediated damage to the FRα-autoantibody complex. Both types can reduce overall folate transport into the brain. FRα autoimmunity is also implicated in CFD syndromes presenting later in childhood, adolescence, or adulthood.
The development of FRα autoimmunity is hypothesized to be linked to cow’s milk consumption in genetically susceptible individuals. The high homology between human FRα and FRα in animal milk may trigger antibody production that cross-reacts with human FRα at the choroid plexus, thyroid, and gonads. Eliminating animal milk from the diet can reduce FRα autoantibody levels, while re-exposure can cause them to rebound. Importantly, an inverse correlation exists between FRα autoantibody titers and CSF MTHF levels, further supporting the pathogenic role of these antibodies in cfd diagnosis. Fluctuations in FRα autoantibody titers over time can also complicate cfd diagnosis and treatment monitoring.
Mitochondrial disorders, including Kearns-Sayre syndrome and Alper’s disease, are another significant cause of CFD. These disorders impair ATP production, which is essential for active folate transport at the choroid plexus and intracellular folate storage. Genetic mutations in FOLR1, the gene encoding FRα, can also directly cause CFD by resulting in loss of FRα function. Capicua transcriptional repressor (CIC) gene variants have also been identified as potential contributors to CFD by downregulating FOLR1 expression.
Inborn errors of folate metabolism within the CNS also play a role in CFD. 3-Phosphoglycerate dehydrogenase deficiency, a disorder of serine synthesis, leads to a deficiency of one-carbon units needed for folate metabolism in the brain. Methenyl-THF synthetase deficiency, dihydropteridine reductase (DHPR) deficiency, and aromatic L-amino acid decarboxylase deficiency are other rare inborn errors that can disrupt folate homeostasis in the CNS and contribute to CFD. Rett syndrome and a variant of Aicardi-Goutières syndrome have also been associated with CFD, although the underlying mechanisms are less clear. Elevated reactive oxygen species (ROS) can also contribute to CFD by promoting MTHF catabolism and impairing FRα and RFC1 function.
6. Clinical Classification of CFD Syndromes Based on Age of Onset
CFD syndromes exhibit age-dependent clinical presentations, particularly those associated with FRα autoantibodies. Infantile CFDS, the classic form, typically emerges in the first three years of life, starting around 4-6 months with agitation, sleep problems, and restlessness. This progresses to neurodevelopmental delay, slowed head growth, hypotonia, ataxia, pyramidal signs, and in some cases, seizures and dyskinesias. Visual and hearing loss can develop later in untreated cases. MRI findings may include delayed myelination and cerebral/cerebellar atrophy. Cfd diagnosis in infantile CFDS relies on CSF folate levels and FRα autoantibody testing, as routine lab tests are usually normal. Early cfd diagnosis and folinic acid treatment are crucial for preventing neurological deterioration and promoting recovery.
A subset of children with infantile autism also exhibit CFD, often with similarly low CSF MTHF and FRα autoantibody levels as those with infantile CFDS. Parental FRα autoimmunity may play a role in autism with CFD, suggesting potential prenatal influences on neurodevelopment. Children with autism and CFD may have slightly higher CSF MTHF levels compared to those with infantile CFDS and neurological deficits, but still lower than healthy controls.
Spastic-ataxic CFD syndrome can develop in children between 1 and 2 years old with postnatal FRα antibody development, leading to learning deficits. In older children (2-5 years), FRα autoimmunity can be associated with attention deficit hyperkinetic disorder (ADHD), learning difficulties, and subtle neurological signs. Adolescent or adult-onset FRα autoimmunity can manifest as severe psychotic episodes and refractory schizophrenia, often with fluctuating FRα antibody titers that correlate with clinical symptom severity. Treatment-resistant major depression and dystonia/Parkinsonism with psychiatric features are other conditions where CFD due to FRα autoimmunity should be considered in the differential cfd diagnosis. Rarely, CFD has been reported in adult dementia with myoclonus.
The specific clinical presentation of CFD due to FRα autoimmunity is influenced by the age of onset of autoantibody development and the presence or absence of parental FRα autoimmunity, particularly in autism spectrum disorders (Figure 4).
Figure 4: Age-Dependent Classification of CFDS. Illustrating the range of CFDS clinical presentations across different age groups, from infantile onset to adulthood, emphasizing the age-related variability in symptoms and associated conditions.
7. Diagnostic Investigations for CFD: A Comprehensive Approach
Cfd diagnosis begins with a thorough patient history, physical examination, and neurological/psychiatric assessments. Brain imaging (MRI), EEG, and neurophysiological studies may be indicated based on clinical suspicion. If CFD is suspected, initial investigations include a complete blood count, serum and red blood cell folate, vitamin B12, homocysteine, and lactate levels, along with metabolic screening. However, the cornerstone of cfd diagnosis is a lumbar puncture to measure CSF glucose, protein, cell count, and crucially, CSF MTHF levels. Analysis of CSF neurotransmitter metabolites (dopamine, serotonin), pterin metabolites (neopterin, biopterin), and potentially lactate and amino acids may also be informative.
Serum FRα autoantibody testing, both blocking and binding types, is essential in suspected CFD, particularly in infantile CFDS and autism. Repeated testing may be necessary due to potential antibody titer fluctuations. It’s important to avoid folate-containing supplements for three days before FRα autoantibody testing. If FRα antibodies are negative, further investigations should consider mitochondrial disorders, FOLR1 and MTHFR gene mutations, and oxidative stress biomarkers, along with assessment of antioxidant vitamin and mineral status.
In infantile CFD or autism, serum FRα autoantibody testing can be a primary screening step, potentially preceding a lumbar puncture in some cases, as positive FRα antibodies strongly suggest CFD and correlate with low CSF MTHF. The specific diagnostic algorithm for cfd diagnosis should be tailored to the individual patient, guided by clinical presentation and history (Figure 5).
Figure 5: Diagnostic Algorithm for Infantile CFD and Autistic Syndromes. Presenting a step-by-step approach to diagnosing CFD in infants and children with autism, starting with clinical suspicion and progressing through serum and CSF testing, and further investigations as needed.
8. Treatment Strategies for Cerebral Folate Deficiency
Once cfd diagnosis is confirmed, prompt treatment is crucial. For CFD due to FRα autoimmunity, high-dose folinic acid (dl-5-formyltetrahydrofolate) is the primary treatment. Typical starting doses are 0.5-1 mg/kg/day, potentially increasing to 2 mg/kg/day (maximum 50 mg/day). Levo-folinic acid or levo-5-methyl-tetrahydrofolate are alternative options. Initiating folinic acid gradually over the first month is recommended to avoid rapid increases in brain folate and neurotransmitter levels, which can cause agitation. If agitation occurs, low-dose risperidone may be used temporarily.
A strict animal-milk-free diet, using plant-based milk alternatives, is a valuable adjunct therapy for CFD due to FRα autoimmunity. Dietary changes can help reduce FRα autoantibody levels over time. However, dietary compliance can be challenging, especially in children with autism. In severe cases of infantile CFDS with dyskinesia, seizures, or status epilepticus, corticosteroids may be used to temporarily suppress FRα autoimmunity while awaiting the effects of folinic acid and dietary changes. Intravenous immunoglobulin (IVIG) has been explored, but its efficacy is not definitively established.
Early treatment with folinic acid in infantile CFDS is associated with better outcomes. Treatment initiation before age two can lead to significant recovery or even complete resolution of symptoms. Early cfd diagnosis is therefore critical. In autism with CFD, early folinic acid and dietary intervention are also important, although parental FRα autoimmunity can influence treatment response and prognosis. If folinic acid therapy is not effective after four months, parental FRα antibody testing and further metabolic/genetic investigations should be considered. For CFD due to FOLR1 mutations, even higher doses of folinic acid (up to 5-7 mg/kg/day) may be required. CFD associated with mitochondrial disorders may show variable response to folinic acid, with antioxidants potentially providing additional benefit.
Addressing co-existing nutritional deficiencies is vital in CFD management, particularly in autism where selective eating and micronutrient deficiencies are common. Vitamin B12, vitamin D, other vitamins, and trace elements should be assessed and supplemented as needed. Vitamin D deficiency correction is important as it can improve RFC1 gene expression and folate transport. Riboflavin supplementation may be beneficial in individuals with MTHFR mutations to enhance MTHFR enzyme activity and MTHF production. In refractory psychotic or schizophrenic syndromes with fluctuating FRα autoantibody titers, folinic acid treatment may help stabilize mood and symptoms. Folinic acid may also be beneficial in major depression associated with FRα autoimmunity.
9. Future Directions and Perspectives in CFD Management
Early cfd diagnosis and intervention are paramount for optimizing outcomes in infantile-onset CFDS and autism with CFD. Raising clinical awareness of these conditions, particularly in young children with unexplained neurological or developmental issues, is crucial. Prognosis may be less favorable when parental FRα autoimmunity is present, highlighting the need for comprehensive family assessment.
For families with a child diagnosed with autism and parental FRα autoimmunity, pre-conception and prenatal folinic acid supplementation for FRα antibody-positive parents may be a promising preventative strategy for future offspring (Figure 6). Genetic counseling is essential to rule out genetic causes of autism before planning subsequent pregnancies. Further research, including whole-exome sequencing studies, is ongoing to identify additional genetic factors contributing to CFD and FRα autoimmunity. Screening for maternal and cord blood FRαAb, and monitoring children for FRαAb in early childhood, could be considered for early cfd diagnosis and intervention. Clinical trials are warranted to further validate the effectiveness of early intervention and preventative strategies for CFDS and ASD.
Figure 6: Genetic Counseling and Preventative Strategies for Recurrence of Autism. (A) Illustrating the role of genetic counseling and FRαAb testing in families with a child with autism to prevent recurrence in subsequent children. (B) Depicting various scenarios of FRα autoantibody presence in children and parents and their implications for recurrence risk and preventative measures.
Author Contributions:
Conceptualization, formal analysis, investigation, data curation, V.T.R.; writing—original draft preparation, writing—review and editing, V.T.R. and E.V.Q.; All authors have read and agreed to the published version of the manuscript.
Institutional Review Board Statement:
Not Applicable.
Informed Consent Statement:
Informed consent was obtained from all subjects reported in this study.
Data Availability Statement:
Not Applicable.
Conflicts of Interest:
The authors declare no conflict of interest.
Funding Statement:
This research received no external funding.
Footnotes:
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References:
[List of references from original article – retain original numbering and links]
Associated Data:
Data Availability Statement:
Not Applicable.