Introduction
Encephalitis, a serious condition marked by brain inflammation, presents a significant diagnostic challenge due to its diverse etiologies and complex clinical presentations. While infections have historically been considered the primary cause, the last decade has witnessed a paradigm shift with the recognition of autoimmune encephalitis (AE) as a major contributor. This advancement is largely attributed to the identification of novel syndromes and biomarkers, particularly antibodies targeting neuronal cell-surface or synaptic proteins, which have revolutionized our understanding and diagnostic strategies for these disorders.
Despite these breakthroughs, current diagnostic criteria for AE often lean heavily on antibody testing and therapeutic responses, potentially causing delays in diagnosis and treatment initiation. This reliance is problematic as antibody tests may not be readily accessible, and their results can take considerable time to process. Moreover, a negative antibody test does not definitively rule out AE, and conversely, a positive result doesn’t always confirm the diagnosis. Similarly, waiting for a response to immunotherapy is not a feasible initial diagnostic step, especially when early intervention is crucial for optimal patient outcomes.
Recognizing these limitations and the critical need for timely diagnosis, this article presents a practical, syndrome-based clinical approach to AE diagnosis. Developed by a team of experts, these guidelines prioritize neurological assessment and conventional diagnostic tests that are widely available to clinicians. By employing a logical differential diagnostic process, we aim to establish levels of evidence for AE – possible, probable, or definite – at the earliest stages of evaluation. This approach facilitates prompt immunotherapy initiation while more specialized investigations, such as comprehensive antibody testing, are underway to refine the diagnosis and guide subsequent treatment strategies. Our objective is to equip clinicians with a robust framework for the initial clinical assessment of suspected AE, ensuring that patients receive timely and appropriate care.
Table 1. Antibodies in the Diagnosis of Autoimmune Encephalitis
| Syndrome | Diagnostic Assay | Frequency of Cancer | Main Type of Cancer |
|---|---|---|---|
| Antibodies against intracellular antigens | |||
| Hu (ANNA1)8* | Limbic encephalitis | >95% | Small-cell lung carcinoma |
| Ma29 | Limbic encephalitis† | >95% | Testicular seminoma |
| GAD10 | Limbic encephalitis‡ | Radioimmunoassay | 25%§ |
| Antibodies against synaptic receptors | |||
| NMDA receptor11 | Anti-NMDA receptor encephalitis | Cell-based assay | Varies with age and sex |
| AMPA receptor12 | Limbic encephalitis | Cell-based assay | 65% |
| GABAB receptor13 | Limbic encephalitis | Cell-based assay | 50% |
| GABAA receptor14 | Encephalitis | Cell-based assay | Thymoma |
| mGluR515 | Encephalitis | Cell-based assay | 70% |
| Dopamine 2 receptor16 | Basal ganglia encephalitis | Cell-based assay | 0% |
| Antibodies against ion channels and other cell-surface proteins | |||
| LGI117 | Limbic encephalitis | Cell-based assay | 5–10% |
| CASPR218 | Morvan’s syndrome|| or limbic encephalitis | Cell-based assay | 20–50% |
| DPPX19 | Encephalitis†† | Cell-based assay | Lymphoma |
| MOG20‡‡ | Acute disseminated encephalomyelitis | Cell-based assay | 0% |
| Aquaporin 421‡‡ | Encephalitis | Cell-based assay | 0% |
| GQ1b22 | Bickerstaff’s brainstem encephalitis | ELISA | 0% |
GAD=glutamic acid decarboxylase. LGI1=leucine-rich glioma inactivated 1. CASPR2=contactin associated protein 2. DPPX=dipeptidyl-peptidase-like protein-6. MOG=myelin oligodendrocyte glycoprotein.
[]Amphiphysin or CV2 (CRMP5) antibodies instead of Hu antibodies in a few patients with limbic encephalitis and small-cell lung carcinoma.*
[†]Limbic encephalitis frequently associated with hypothalamic and mesencephalic involvement.
[‡]GAD antibodies occur more frequently in patients with stiff person syndrome and cerebellar ataxia. The association with cancer preferentially occurs in patients with limbic encephalitis.
[§]Tumours found more frequently in men older than 50 years.23
[¶]Ovarian teratoma usually found in young women aged 12–45 years.
[||]Morvan’s syndrome usually has a more chronic clinical course, but might present with predominant cognitive and behavioural symptoms fulfilling criteria of possible autoimmune encephalitis.
*[*]Thymoma associated with Morvan’s syndrome rather than limbic encephalitis.
[††]Encephalitis associated with diarrhoea and hyperekplexia.
[‡‡]Mostly restricted to children.
Scope and Objectives of these Guidelines
These guidelines are specifically designed to address autoimmune encephalitis presenting with a subacute onset of memory deficits or altered mental status. These core symptoms may be accompanied by a range of other neurological and psychiatric manifestations. Our primary goal is to facilitate a rapid and accurate diagnosis using a clinically focused approach. It is important to note that these guidelines are not intended for other central nervous system (CNS) autoimmune disorders such as stiff person syndrome, progressive encephalomyelitis with rigidity and myoclonus, or autoimmune cerebellopathies, which typically exhibit distinct clinical profiles.
The existing diagnostic landscape for AE often places undue emphasis on antibody test results and response to immunotherapy. We argue against this over-reliance, particularly in the initial diagnostic phase. Antibody testing, while crucial for definitive diagnosis and subtype characterization, is not universally accessible, and result turnaround times can be lengthy. Moreover, the absence of detectable autoantibodies does not negate the possibility of an immune-mediated etiology. Conversely, a positive antibody test must be interpreted cautiously within the clinical context, as it does not automatically confirm a definitive diagnosis of AE. Similarly, using immunotherapy response as a primary diagnostic criterion is impractical for initial assessments. Early immunotherapy has been shown to improve outcomes in AE, and delaying treatment while awaiting antibody results or observing treatment response can be detrimental.
Therefore, these guidelines advocate for an initial diagnostic approach grounded in conventional neurological evaluation and readily available standard diagnostic tests, such as MRI, CSF analysis, and EEG. This strategy enables clinicians to initiate preliminary treatment promptly while awaiting comprehensive antibody test results. Subsequent antibody findings then serve to refine the diagnosis, guide long-term management, and tailor therapeutic strategies.
The focus of these guidelines and the emphasis on initial clinical assessment explain the inclusion or exclusion of certain disorders. For instance, acute disseminated encephalomyelitis (ADEM) is included due to its clinical overlap with other autoimmune encephalitis entities. Hashimoto’s encephalopathy, while its existence is debated, is also addressed because it frequently appears in the differential diagnosis of AE. Conversely, Morvan’s syndrome and Rasmussen’s encephalitis, although autoimmune in nature, are discussed in the appendix due to their more chronic course and differing initial symptom presentations. We acknowledge the potential overlap between these conditions and AE, hence their consideration in the supplementary material.
Given that the spectrum of autoimmune encephalitis in children differs from adults, and that pediatric presentations can be less typical, these guidelines should be applied judiciously in children, especially those under 5 years of age. Pediatric AE often requires a nuanced diagnostic approach, and these guidelines should be adapted accordingly in younger populations.
Methodology
These guidelines were developed through a rigorous, multi-stage process. Initially, two authors (FG and JD) drafted the guidelines, which then underwent three rounds of review and refinement by a panel of experts in autoimmune encephalitis. The first stage involved a thorough review of existing encephalitis diagnostic criteria and guidelines (encompassing both infectious and idiopathic etiologies). This review, combined with extensive clinical experience in diagnosing and managing newly recognized forms of AE (including those presenting with subtle cognitive or behavioral changes without overt consciousness alteration), led to the formulation of criteria for “possible autoimmune encephalitis.” Crucially, these initial criteria are independent of neuronal autoantibody status, recognizing the limitations of relying solely on antibody testing for early diagnosis.
Next, we critically evaluated existing diagnostic criteria for specific clinical syndromes associated with AE, such as limbic encephalitis and Bickerstaff’s brainstem encephalitis. We identified areas where criteria were lacking clarity or needed modification to better reflect the current understanding of AE. We then refined existing criteria and developed new diagnostic criteria, for example, for probable anti-NMDA receptor encephalitis. These revised and novel criteria emphasize symptom assessment and standard paraclinical tests, minimizing reliance on autoantibody status for initial diagnostic categorization.
This iterative process resulted in the establishment of a three-tiered system of clinical evidence for AE: possible, probable, and definite. The “possible” and “probable” levels are designed to facilitate early clinical diagnosis and treatment initiation, often without requiring autoantibody confirmation. The “definite” level typically incorporates autoantibody status for diagnostic confirmation and subtype classification. Concurrently, we conducted a comprehensive review of the literature and our collective experience with neuronal autoantibody studies. This review aimed to identify potential pitfalls in antibody test interpretation, leading to the development of practical recommendations for the appropriate utilization and interpretation of autoantibody findings in the diagnostic workup of AE.
Initial Clinical Assessment: Possible Autoimmune Encephalitis
The concept of “possible autoimmune encephalitis” is introduced to facilitate early recognition and prompt initiation of investigations and potential treatment. A patient presenting with new-onset encephalitis should be considered to have possible AE if they meet all three criteria outlined in Panel 1. These criteria represent a departure from traditional encephalitis diagnostic approaches, which often emphasize altered consciousness, fever, CSF pleocytosis, and EEG abnormalities. The criteria for possible AE are intentionally broader to encompass the diverse clinical spectrum of autoimmune encephalitis, where patients may present with prominent memory or behavioral deficits, subtle changes in consciousness, or even normal findings on initial brain MRI or CSF analysis. In this context, “memory deficits” specifically refer to impairments in forming new long-term memories due to hippocampal dysfunction, as well as challenges with working memory, the system responsible for temporary information storage and manipulation.
Panel 1. Diagnostic Criteria for Possible Autoimmune Encephalitis.
Diagnosis can be made when all three of the following criteria have been met:
- Subacute onset (rapid progression over less than 3 months) of one or more of the following:[*]
- Memory deficits
- Altered mental status
- Psychiatric symptoms
- At least one of the following new findings:[†]
- Focal neurological deficits
- Seizures not explained by a prior seizure disorder
- CSF pleocytosis
- MRI features suggestive of autoimmune encephalitis[†]
- Reasonable exclusion of alternative causes.
[] Altered mental status defined as decreased or altered level of consciousness, lethargy, or personality change.*
[†] Brain MRI hyperintense signal on T2-weighted fluid-attenuated inversion recovery sequences highly restricted to one or both medial temporal lobes (limbic encephalitis), or in multifocal areas involving grey matter, white matter, or both compatible with demyelination or inflammation.
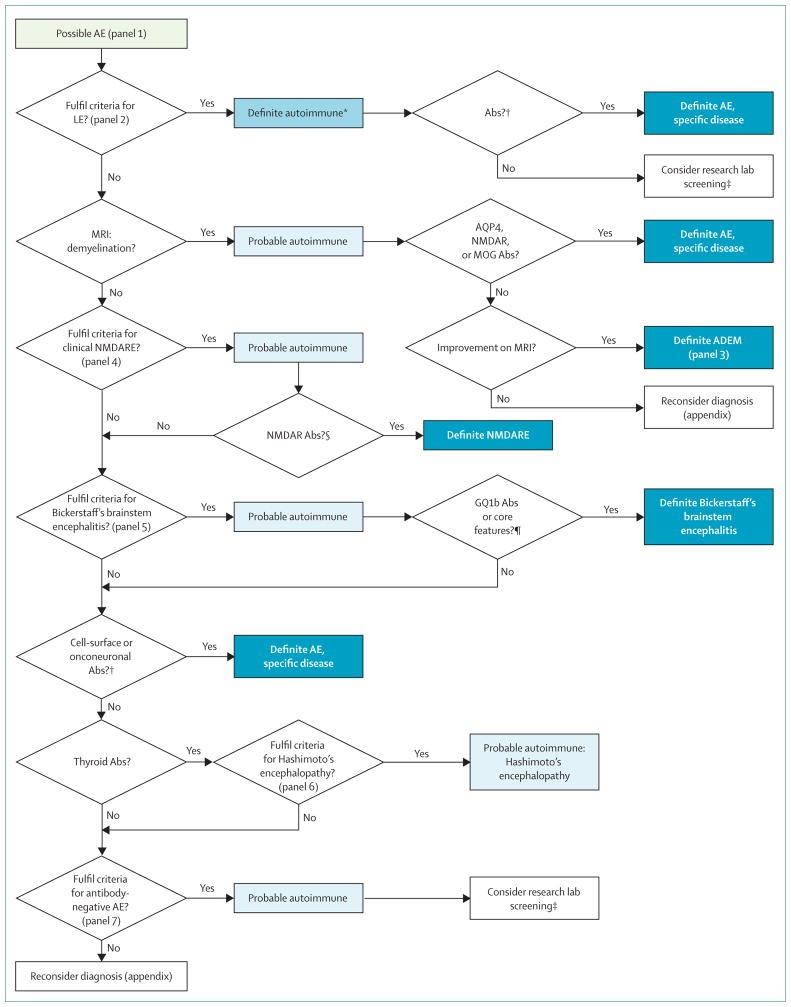
Figure 1. Algorithm for the diagnosis of autoimmune encephalitis.
Brain MRI is a crucial component of the initial evaluation for encephalitis. While findings may be normal or non-specific in early stages of AE, certain patterns can raise suspicion for an autoimmune etiology. For instance, bilateral medial temporal lobe involvement is suggestive of limbic encephalitis, and multifocal white matter lesions may indicate ADEM or other demyelinating forms of AE. In contrast, EEG alterations are generally less specific for AE. However, specific EEG patterns, such as extreme delta brush in anti-NMDA receptor encephalitis, can be diagnostically helpful. EEG is also valuable in excluding other conditions like Creutzfeldt-Jakob disease and in detecting subclinical seizures or non-convulsive status epilepticus, which are common in AE.
In addition to meeting the criteria for possible AE, a thorough differential diagnosis is essential. Clinicians must carefully evaluate and exclude other conditions that can mimic AE and cause rapidly progressive encephalopathy. The appendix provides a detailed discussion of differential diagnoses. In most cases, a detailed clinical history, comprehensive general and neurological examination, routine blood and CSF analysis, and brain MRI, including diffusion-weighted imaging, will suffice to rule out common mimics. Herpes simplex virus encephalitis and other CNS infections are particularly important to exclude. It’s crucial to remember that CSF herpes simplex virus PCR may be falsely negative if performed too early in the disease course (e.g., within the first 24 hours). If clinical suspicion for herpes simplex virus encephalitis remains high, repeat PCR testing is warranted. Prior reviews have extensively covered the differential diagnosis of infectious encephalitis, and clinicians should consult these resources for further guidance.
Clinical Syndromes Suggestive of Autoimmune Encephalitis
While some patients with autoimmune encephalitis do not present with clearly defined syndromes, a significant proportion exhibit recognizable clinical patterns. In some instances, demographic factors or specific comorbidities (e.g., diarrhea, ovarian teratoma, faciobrachial dystonic seizures) can provide initial clues towards the underlying antibody-mediated disorder (e.g., anti-DPPX, anti-NMDA receptor, anti-LGI1 encephalitis, respectively). However, these features are not pathognomonic and may be absent in some patients. In such cases, the diagnosis of definite autoimmune encephalitis often hinges on autoantibody test results.
Conversely, certain disorders are characterized by distinct clinical syndromes and MRI findings that allow for classification as probable or even definite autoimmune encephalitis even before autoantibody status is known. These include limbic encephalitis, acute disseminated encephalomyelitis, syndromes with predominantly white matter MRI features, anti-NMDA receptor encephalitis, and Bickerstaff’s brainstem encephalitis (Figure 1). These syndromes are discussed in detail below, highlighting the clinical and paraclinical features that aid in their diagnosis.
Autoimmune Limbic Encephalitis
Autoimmune limbic encephalitis (LE) is a well-recognized syndrome characterized by subacute onset of symptoms indicating limbic system dysfunction. The diagnostic criteria for definite autoimmune LE are presented in Panel 2. These criteria are modified from previous versions to incorporate the presence of bilateral medial temporal lobe involvement on T2-weighted FLAIR MRI (Figure 2). Importantly, in these proposed criteria, antibody status is not mandatory for diagnosing definite autoimmune LE. Immune-mediated LE can occur without detectable autoantibodies, emphasizing the importance of clinical and MRI findings in diagnosis.
Panel 2. Diagnostic Criteria for Definite Autoimmune Limbic Encephalitis.
Diagnosis can be made when *all four[]** of the following criteria have been met:
- Subacute onset (rapid progression of less than 3 months) of working memory deficits, seizures, or psychiatric symptoms suggesting involvement of the limbic system.
- Bilateral brain abnormalities on T2-weighted fluid-attenuated inversion recovery MRI highly restricted to the medial temporal lobes[†].
- At least one of the following:
- CSF pleocytosis (white blood cell count of more than five cells per mm3)
- EEG with epileptic or slow-wave activity involving the temporal lobes
- Reasonable exclusion of alternative causes (appendix).
[] If one of the first three criteria is not met, a diagnosis of definite limbic encephalitis can be made only with the detection of antibodies against cell-surface, synaptic, or onconeural proteins.*
[†] 18Fluorodeoxyglucose (18F-FDG) PET can be used to fulfil this criterion. Results from studies from the past 5 years suggest that 18F-FDG-PET imaging might be more sensitive than MRI to show an increase in FDG uptake in normal-appearing medial temporal lobes.44,45
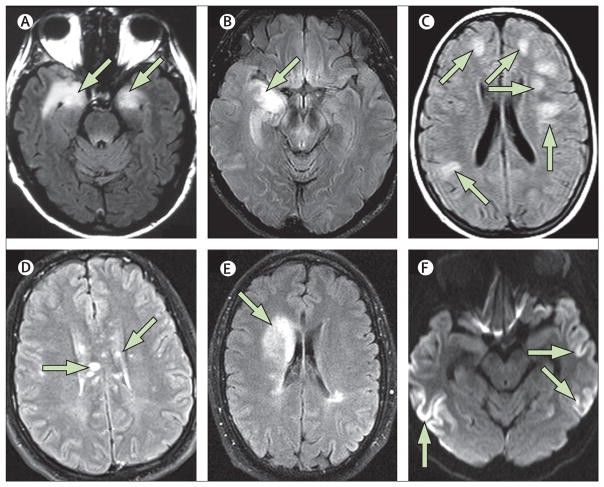
Figure 2. MRI patterns in autoimmune encephalitis and its mimics.
Measuring autoantibodies in suspected LE remains crucial for several reasons. First, antibody identification clarifies the immunological subtype of LE, which has implications for prognosis, tumor association, comorbidity risks, and treatment strategies. Different antibodies are associated with varying clinical courses and therapeutic responses. Second, in patients who do not fully meet the clinical criteria for definite LE, detecting specific autoantibodies can establish the diagnosis of autoimmune LE. Thus, while not essential for initial diagnosis of definite LE when clinical and MRI criteria are met, antibody testing plays a vital role in refining the diagnosis and guiding long-term management.
The hallmark clinical presentation of LE is the rapid development of confusion, working memory deficits, mood changes, and often seizures. Subacute onset of short-term memory loss is a particularly characteristic feature, although it may be overshadowed by other prominent symptoms. CSF analysis in LE typically reveals mild-to-moderate lymphocytic pleocytosis (usually <100 white blood cells/mm3) in 60-80% of patients. Elevated IgG index or oligoclonal bands are found in approximately 50% of cases, indicating intrathecal immunoglobulin synthesis. However, patients with LGI1 antibody-associated LE may have a lower frequency of CSF pleocytosis (41%) or elevated CSF protein concentrations (47%), and rarely exhibit intrathecal IgG synthesis. The relative absence of inflammatory CSF findings in LGI1-antibody LE can initially suggest a non-inflammatory encephalopathy, highlighting the importance of considering antibody testing even with seemingly non-inflammatory CSF profiles.
MRI in LE often demonstrates increased T2-weighted FLAIR signal in the medial temporal lobes bilaterally. While LE can present with unilateral MRI involvement or even a normal MRI, these cases are not classified as definite LE according to our criteria unless specific antibodies are subsequently detected. This is because unilateral medial temporal lobe MRI abnormalities can be caused by various non-immune conditions, including seizures, herpes simplex virus encephalitis, and gliomas. Distinguishing LE from these mimics requires careful clinical and radiological evaluation. For instance, MRI findings in immunocompromised patients with human herpes virus 6-associated encephalitis can closely resemble autoimmune LE, but the clinical context is typically different. Herpes simplex virus encephalitis, in contrast, tends to have more widespread involvement beyond the limbic system, may exhibit hemorrhagic features, and often shows restricted diffusion and contrast enhancement.
Certain demographic and clinical clues can suggest the underlying immune etiology in LE. For example, LE associated with onconeural antibodies (e.g., Hu, Ma2) is strongly suggestive of an underlying malignancy. However, definitive immunological subtyping of LE relies on autoantibody measurement. Distinguishing between immunological subtypes is crucial because LE associated with onconeural antibodies generally responds less favorably to immunotherapy compared to LE associated with cell-surface antibodies. The onconeural antibodies most commonly associated with LE are Hu and Ma2, and their presence almost invariably indicates an underlying cancer. Conversely, the neuronal cell-surface antibodies most frequently linked to LE include LGI1, GABAB receptor, and AMPA receptor antibodies. The frequency and type of tumors associated with different antibodies are summarized in Table 1.
Antibodies against glutamic acid decarboxylase (GAD), an intracellular antigen, are found in a subgroup of LE patients, predominantly young women (median age 23 years) with seizures as a prominent feature and no evidence of cancer. However, the risk of malignancy, typically small-cell lung carcinoma or thymoma, is elevated in patients with GAD antibodies and LE who are older than 50 years or have concurrent GABAB receptor antibodies.
Acute Disseminated Encephalomyelitis (ADEM) and Demyelinating Syndromes
Acute disseminated encephalomyelitis (ADEM) is an inflammatory demyelinating disorder of the CNS, typically monophasic, that predominantly affects children and young adults under 40 years of age. ADEM often follows an acute systemic infection or vaccination. Encephalopathy, of variable severity, is a mandatory diagnostic criterion for definite ADEM (Panel 3). Other neurological signs, such as cranial nerve palsies, ataxia, hemiparesis, myelopathy, and optic neuritis, may also be present. CSF analysis in ADEM typically shows mild pleocytosis (<50 lymphocytes/mm3), while CSF oligoclonal bands are uncommon (<7% of cases). Brain MRI in ADEM reveals multiple, large (>2 cm) T2-weighted FLAIR abnormalities, which can be located in supratentorial white matter, basal ganglia, brainstem, cerebellum, and spinal cord. Contrast enhancement may or may not be present. Currently, there are no specific biomarkers for ADEM. Diagnostic criteria for definite ADEM have been proposed for children (Panel 3).
Panel 3. Diagnostic Criteria for Definite Acute Disseminated Encephalomyelitis32.
Diagnosis can be made when all five of the following criteria have been met:
- A first multifocal, clinical CNS event of presumed inflammatory demyelinating cause.
- Encephalopathy that cannot be explained by fever.
- Abnormal brain MRI:
- Diffuse, poorly demarcated, large (>1–2 cm) lesions predominantly involving the cerebral white matter.
- T1-hypointense lesions in the white matter in rare cases.
- Deep grey matter abnormalities (e.g., thalamus or basal ganglia) can be present.
- No new clinical or MRI findings after 3 months of symptom onset.
- Reasonable exclusion of alternative causes.
Figure 2. MRI patterns in autoimmune encephalitis and its mimics.
According to these criteria, a key requirement for definite ADEM is the absence of new clinical or MRI findings 3 months after symptom onset, reflecting its monophasic nature. While this criterion is inherently retrospective, the remaining criteria are robust enough to establish a diagnosis of probable ADEM at presentation, allowing for prompt immunotherapy initiation.
Myelin oligodendrocyte glycoprotein (MOG) antibodies may be transiently present in approximately 50% of children with ADEM. However, MOG antibody testing is not currently incorporated into ADEM diagnostic criteria. This is because MOG antibodies can be found in demyelinating disorders with encephalopathy but without typical ADEM MRI features, as well as in demyelinating disorders without encephalopathy. Furthermore, MOG antibody testing remains unavailable in many centers.
Susac’s syndrome is an important, albeit rare, differential diagnosis to consider in patients meeting criteria for possible AE with demyelinating MRI features. Susac’s syndrome is thought to be an autoimmune vasculopathy causing microvessel thromboses affecting the brain, retina, and inner ear. Encephalopathy is present in a majority (76%) of Susac’s syndrome cases. However, simultaneous involvement of all three levels (brain, retina, inner ear) at disease onset is relatively uncommon, occurring in only 13% of cases. Diagnosis of Susac’s syndrome is based on the presence of branch retinal artery occlusions detected by fluorescein angiography and characteristic MRI findings, including snowball-like lesions or “holes” in the central corpus callosum, along with other periventricular white matter abnormalities on T2-weighted FLAIR imaging. These MRI features differ from those typically seen in ADEM and are highly suggestive of Susac’s syndrome in the context of encephalopathy.
Anti-NMDA Receptor Encephalitis
Anti-NMDA receptor encephalitis is frequently recognizable based on its distinctive clinical presentation and is associated with CSF IgG antibodies against the GluN1 subunit of the NMDA receptor. These antibodies are highly specific and have demonstrated pathogenicity. This disorder predominantly affects young individuals, with 95% of cases occurring in those under 45 years of age, and a female predominance of 4:1 overall. The female predominance is less pronounced in young children (<12 years) and older adults (>45 years). The frequency of underlying tumors, particularly ovarian teratomas in women, varies with age and sex. Ovarian teratomas are found in up to 58% of women over 18 years, but are rare in children. In older adults (>45 years), tumors are less frequent (23%) and are more likely to be carcinomas rather than teratomas.
Teenagers and adults with anti-NMDA receptor encephalitis typically present with a characteristic sequence of symptoms. Initial symptoms often include abnormal behavior (psychosis, delusions, hallucinations, agitation, aggression, catatonia), irritability, and insomnia. These are followed by speech dysfunction, dyskinesias, memory deficits, autonomic instability, and decreased level of consciousness. Seizures can occur at any point in the disease course, but tend to be earlier in males. Young children more commonly present with abnormal movements or seizures. However, regardless of age, the clinical picture tends to converge by 3-4 weeks after symptom onset. By the end of the first month, most patients exhibit a combination of symptoms across multiple domains, including behavioral and cognitive abnormalities, memory deficits, speech disorder, seizures, abnormal movements, altered consciousness or autonomic dysfunction, central hypoventilation, and cerebellar ataxia or hemiparesis. Isolated symptom presentations are rare.
Based on these clinical features, and while awaiting confirmatory IgG anti-GluN1 antibody results, a patient with rapidly progressive encephalopathy can be considered to have probable anti-NMDA receptor encephalitis if they meet the criteria in Panel 4. Memory deficit, while common, is not included as a mandatory criterion due to the difficulty in assessing it in patients with psychosis, agitation, or in young children. Similarly, hemiparesis and cerebellar ataxia are less frequent and are therefore not core criteria. In patients meeting these criteria, immunotherapy and investigation for an underlying neoplasm (guided by age and sex) should be initiated promptly. Retrospective analysis of a large cohort study demonstrated that 80% of patients with confirmed anti-NMDA receptor encephalitis met these probable criteria within the first month of symptom onset.
Panel 4. Diagnostic Criteria for Anti-NMDA Receptor Encephalitis.
Probable Anti-NMDA Receptor Encephalitis[*]
Diagnosis can be made when all three of the following criteria have been met:
- Rapid onset (less than 3 months) of at least four of the six following major groups of symptoms:
- Abnormal (psychiatric) behavior or cognitive dysfunction
- Speech dysfunction (pressured speech, verbal reduction, mutism)
- Seizures
- Movement disorder, dyskinesias, or rigidity/abnormal postures
- Decreased level of consciousness
- Autonomic dysfunction or central hypoventilation
- At least one of the following laboratory study results:
- Abnormal EEG (focal or diffuse slow or disorganized activity, epileptic activity, or extreme delta brush)
- CSF with pleocytosis or oligoclonal bands
- Reasonable exclusion of other disorders (appendix)
Diagnosis can also be made in the presence of three of the above groups of symptoms accompanied by a systemic teratoma.
Definite Anti-NMDA Receptor Encephalitis[*]
Diagnosis can be made in the presence of one or more of the six major groups of symptoms and IgG anti-GluN1 antibodies,[†] after reasonable exclusion of other disorders (appendix).
[] Patients with a history of herpes simplex virus encephalitis in the previous weeks might have relapsing immune-mediated neurological symptoms (post-herpes simplex virus encephalitis).*
[†] Antibody testing should include testing of CSF. If only serum is available, confirmatory tests should be included (e.g., live neurons or tissue immunohistochemistry, in addition to cell-based assay).
Figure 1. Algorithm for the diagnosis of autoimmune encephalitis.
Patients with partial symptom presentations who may not meet these initial criteria will be identified through antibody testing. Antibody studies should ideally include CSF analysis, as serum-only testing may lead to false-negative or false-positive results. Studies have shown that serum testing can be less reliable and may detect antibodies in patients without anti-NMDA receptor encephalitis or other immune-mediated disorders.
CSF analysis for NMDA receptor antibodies is particularly crucial in patients with relapsing symptoms following herpes simplex encephalitis. This relapsing form of herpes simplex encephalitis is an autoimmune disorder that can be clinically indistinguishable from classic anti-NMDA receptor encephalitis. It affects approximately 20% of patients with herpes simplex encephalitis and manifests with new-onset choreoathetosis (predominantly in children) or psychiatric symptoms (mainly in adults and teenagers) weeks or, rarely, months after the viral infection. In addition to NMDA receptor antibodies, some patients may develop GABAA receptor or dopamine receptor 2 antibodies in this post-herpes simplex encephalitis setting.
Bickerstaff’s Brainstem Encephalitis
Bickerstaff’s brainstem encephalitis (BBE) is characterized by subacute onset (within 4 weeks) of progressive impairment of consciousness, ataxia, and bilateral ophthalmoparesis, typically symmetrical. BBE is often preceded by an infectious event, follows a monophasic course, and generally has a favorable outcome. Additional common features include pupillary abnormalities, bilateral facial palsy, Babinski’s sign, and bulbar palsy. Generalized limb weakness may occur, overlapping with Guillain-Barré syndrome features. CSF pleocytosis is present in approximately 45% of patients. Brain MRI is usually normal, although brainstem abnormalities on T2-weighted FLAIR imaging are seen in about 23% of cases.
Most diagnostic criteria for BBE include the classic triad of altered mental status, bilateral external ophthalmoplegia, and ataxia (Panel 5). IgG anti-GQ1b antibodies are highly specific for BBE and the related Miller-Fisher syndrome. Some clinicians group these disorders under the term “GQ1b antibody syndrome.” The criteria proposed in 2014 do not mandate GQ1b antibody testing for a definitive BBE diagnosis, as up to 32% of patients may not have detectable antibodies. However, GQ1b antibody measurement can confirm the diagnosis in patients with incomplete syndromes, atypical symptoms, or when altered mental status hinders ataxia assessment. The diagnostic complexity of BBE is illustrated by a historical case where a patient initially diagnosed with BBE later developed seizures, hyperthermia, psychosis, and fluctuating manic/catatonic states, potentially suggesting overlap with other autoimmune encephalitis entities like anti-NMDA receptor encephalitis. In such complex cases, testing for both GQ1b and NMDA receptor antibodies (if clinically available) could aid in clarifying the diagnosis.
Panel 5. Diagnostic Criteria for Bickerstaff’s Brainstem Encephalitis.
Probable Bickerstaff’s Brainstem Encephalitis
Diagnosis can be made when both of the following criteria have been met:
- Subacute onset (rapid progression of less than 4 weeks) of all the following symptoms:
- Decreased level of consciousness
- Bilateral external ophthalmoplegia
- Ataxia
- Reasonable exclusion of alternative causes.
Definite Bickerstaff’s Brainstem Encephalitis
Diagnosis can be made in the presence of positive IgG anti-GQ1b antibodies even if bilateral external ophthalmoplegia is not complete or ataxia cannot be assessed, or if recovery has occurred within 12 weeks after onset.
Figure 1. Algorithm for the diagnosis of autoimmune encephalitis.
Differential diagnoses for BBE include Listeria rhombencephalitis, EV71 encephalitis in children, paraneoplastic and postinfectious brainstem encephalitis, CLIPPERS (chronic lymphocytic inflammation with pontine perivascular enhancement responsive to steroids), neurosarcoidosis, and primary CNS lymphoma. Careful clinical and radiological evaluation is crucial to differentiate BBE from these mimics.
Antibody Testing in Autoimmune Encephalitis: Clinical Considerations and Caveats
Detecting specific autoantibodies (Table 1, Figure 1) is critical for establishing a definitive diagnosis of autoimmune encephalitis, identifying immunological subtypes of limbic encephalitis, and aiding in the differential diagnosis of atypical cases. Therefore, antibody measurement is a cornerstone of AE diagnosis. However, clinicians must be aware of potential pitfalls in result interpretation.
Several concepts relevant to classic onconeuronal or GAD antibodies do not apply to antibodies against neuronal cell-surface proteins. Onconeuronal and GAD antibodies target intracellular proteins, are present in both serum and CSF, and their epitopes are linear, allowing for detection using various techniques like ELISA, immunoblotting, and immunohistochemistry. In contrast, antibodies against neuronal cell-surface proteins have distinct properties that influence test selection and result interpretation.
Conformational Antigens
Most antibodies targeting neuronal cell-surface proteins recognize epitopes only when they are in their native conformation. Techniques that preserve native conformation are essential for accurate detection. Cell-based assays, widely used in clinical labs, meet this requirement. Immunohistochemistry on brain sections adapted for membrane proteins and immunocytochemistry of live rodent hippocampal neuron cultures (primarily used in research settings) are also suitable techniques. Standard ELISA or Western blot assays, which may denature proteins, may not reliably detect these conformational epitopes.
Molecular Precision
Target antigens for autoantibodies can be composed of multiple subunits. Antibodies against different subunits may have varying clinical significance. For example, the NMDA receptor is a heterotetramer with two GluN1 and two GluN2/3 subunits. IgG antibodies against the GluN1 subunit are specific for anti-NMDA receptor encephalitis. However, antibodies against linear epitopes of GluN2 or GluR ε2 have been reported in diverse disorders and their clinical relevance is uncertain. Therefore, assays should specifically target the clinically relevant subunit (e.g., GluN1 for NMDA receptor encephalitis).
Molecular precision is also crucial for voltage-gated potassium channel complex (VGKC) antibodies. Initial assays targeted the VGKC itself. However, it was later discovered that the true targets are proteins complexed with VGKC, specifically LGI1 and contactin-associated protein-like 2 (CASPR2). Antibodies against LGI1 and CASPR2 are associated with well-defined syndromes. In contrast, VGKC complex antibodies that do not target LGI1 or CASPR2 are not syndrome-specific and should not be used as definitive evidence of immune-mediated pathogenesis. Therefore, current assays should specifically identify LGI1 and CASPR2 antibodies, rather than generic VGKC complex antibodies.
Immunoglobulin Class
The antibodies associated with autoimmune encephalitis in Table 1 are predominantly IgG antibodies. The clinical significance of detecting IgA or IgM antibodies against these antigens is unclear. For example, while IgG antibodies against the NMDA receptor GluN1 subunit are highly specific for anti-NMDA receptor encephalitis, IgM or IgA antibodies have been reported in serum of patients with various disorders and even in healthy individuals, limiting their diagnostic utility. Therefore, assays should specifically detect IgG antibodies to ensure clinical relevance.
Cerebrospinal Fluid (CSF) Studies
CSF analysis is integral to encephalitis diagnosis, including both infectious and autoimmune etiologies. CSF antibody testing is particularly important in suspected AE for several reasons:
- Increased Sensitivity: CSF antibody detection is often more sensitive than serum testing in AE. Many patients with AE have CSF antibodies, and relevant antibodies may be exclusively found in CSF. For example, in anti-NMDA receptor encephalitis, up to 14% of patients have antibodies in CSF but not in serum.
- Distinct Antibody Repertoire: The antibody profile in CSF and serum can differ in the same patient. For instance, a patient might have NMDA receptor antibodies in both CSF and serum, but GABAA receptor antibodies only in serum. The antibodies present in CSF often more accurately reflect the clinical picture in such cases.
- Clinical Correlation: For certain disorders, like anti-NMDA receptor encephalitis, CSF antibody concentrations correlate better with clinical course and disease severity than serum antibody titers. CSF antibody titers may be a more reliable marker of disease activity.
- Reduced False Positives/Negatives: Neuronal antibody testing using serum and cell-based assays can be prone to false-positive or false-negative results. This issue is less frequent with CSF analysis, potentially due to reduced interference from non-specific antibodies or better reflection of CNS-specific immune responses.
Based on these considerations, it is recommended to include both CSF and serum for neuronal antibody testing in suspected autoimmune encephalitis. An approach of initially testing serum and proceeding to CSF only if serum is negative may delay diagnosis. If serum testing is positive but CSF is negative, or if the clinical presentation is inconsistent with the detected antibody, the possibility of a false-positive serum result or a laboratory error should be considered. In such cases, repeat testing, confirmatory assays (e.g., brain immunohistochemistry, cultured neurons), or contacting the laboratory for further clarification are advisable.
Finally, treatment decisions during the disease course should primarily be guided by clinical assessment rather than solely relying on antibody titers. While antibody titers may correlate with clinical course, this correlation is imperfect, and antibodies often remain detectable even after clinical recovery. Therefore, clinical improvement and neurological examination should be the primary determinants of treatment response and duration.
Antibodies in Demyelinating Disorders Overlapping with Anti-NMDA Receptor Encephalitis
Approximately 4% of patients with anti-NMDA receptor encephalitis may develop overlapping syndromes involving demyelination, either concurrently or sequentially. These overlapping syndromes typically involve co-occurrence of anti-NMDA receptor encephalitis with MOG-related or aquaporin-4 (AQP4)-related syndromes. Clinicians should be aware that demyelinating disorders can present with encephalitis-like features, and overlapping syndromes can occur. Patients with demyelinating disorders presenting with atypical features (e.g., dyskinesias, prominent psychiatric manifestations) or patients with anti-NMDA receptor encephalitis with atypical features (e.g., optic neuritis, demyelination on MRI) should be comprehensively evaluated for coexisting disorders rather than simply attributing the atypical features to an expanded spectrum of a single disease. This evaluation should include testing for AQP4 and MOG antibodies in serum (intrathecal production of these antibodies is rare) and NMDA receptor antibodies in both serum and CSF. Recognizing and diagnosing overlapping syndromes is crucial for tailoring appropriate immunotherapy and management strategies.
GAD Antibodies in Limbic Encephalitis and Other Syndromes
Serum antibodies against intracellular glutamic acid decarboxylase (GAD) can be detected at low titers in approximately 1% of healthy individuals and in a much higher proportion (80%) of patients with type 1 diabetes mellitus. Only high titers of serum GAD antibodies are clinically relevant and associated with autoimmune neurological disorders, such as limbic encephalitis and other syndromes. The definition of “high titer” varies depending on the assay technique, but neurological symptoms typically manifest with titers 100–1000 times higher than those observed in individuals with diabetes.
When evaluating a patient with limbic encephalitis, clinicians should consider that high titers of serum GAD antibodies, although rare, may indicate underlying diabetes or other endocrine disorders. In this context, evidence of intrathecal GAD antibody production or CSF oligoclonal bands strengthens the association with the neurological syndrome and supports a diagnosis of autoimmune GAD-related limbic encephalitis. Conversely, low-titer serum GAD antibodies in the absence of intrathecal synthesis should be interpreted cautiously and may not be directly causative of the neurological syndrome.
Approach to Patients without Recognizable Syndromes or Autoantibodies
After excluding well-defined autoimmune encephalitis syndromes (with or without autoantibodies) and other syndromes associated with established autoantibodies, a subset of patients will remain who meet criteria for possible autoimmune encephalitis (Panel 1) but lack a definitive diagnosis. Patients in this group can be considered to have probable autoimmune encephalitis if they fulfill criteria for Hashimoto’s encephalopathy (Panel 6) or the criteria proposed in Panel 7 for antibody-negative probable AE.
Panel 6. Diagnostic Criteria for Hashimoto’s Encephalopathy.
Diagnosis can be made when all six of the following criteria have been met:
- Encephalopathy with seizures, myoclonus, hallucinations, or stroke-like episodes.
- Subclinical or mild overt thyroid disease (usually hypothyroidism).
- Brain MRI normal or with non-specific abnormalities.
- *Presence of serum thyroid (thyroid peroxidase, thyroglobulin) antibodies[].**
- Absence of well-characterized neuronal antibodies in serum and CSF.
- Reasonable exclusion of alternative causes.
[] There is no disease-specific cutoff value for these antibodies (detectable in 13% of healthy individuals).100*
Figure 1. Algorithm for the diagnosis of autoimmune encephalitis.
Hashimoto’s encephalopathy is a controversial entity, often defined retrospectively by a favorable response to steroids. Despite the name, the pathophysiology remains unclear, and the original case report did not describe steroid responsiveness. Hashimoto’s encephalopathy predominantly affects women across a wide age range. Most patients have overt or subclinical thyroid disease, typically hypothyroidism. By definition, patients develop encephalopathy, which may be associated with seizures, myoclonus, hallucinations, and stroke-like episodes. CSF and brain MRI findings are usually normal or non-specific. Most reported patients improve with corticosteroids, often combined with levothyroxine. However, this favorable outcome may be inherent to the definition of the disorder, which has also been termed “steroid-responsive encephalopathy with autoimmune thyroiditis.”
Patients presenting with non-specific encephalopathy, subclinical or overt thyroid disease, anti-thyroid antibodies, and no better explanation for their symptoms may be considered for a steroid trial. However, thyroid antibodies are not specific for Hashimoto’s encephalopathy, as they are present in a significant proportion of healthy individuals and patients with other autoimmune encephalitis disorders. Similarly, alpha-enolase antibodies have been identified in some Hashimoto’s encephalopathy patients, but they lack specificity and are not reliable biomarkers.
The term “Hashimoto’s encephalopathy” should be reserved for cases where rigorous clinical assessment and comprehensive testing for well-characterized neuronal antibodies have excluded other potential causes of encephalopathy. Given the unclear pathogenesis, Hashimoto’s encephalopathy should be classified as probable autoimmune encephalitis.
Panel 7. Criteria for Autoantibody-Negative but Probable Autoimmune Encephalitis.
Diagnosis can be made when all four of the following criteria have been met:
- Rapid progression (less than 3 months) of working memory deficits (short-term memory loss), altered mental status, or psychiatric symptoms.
- Exclusion of well-defined syndromes of autoimmune encephalitis (e.g., typical limbic encephalitis, Bickerstaff’s brainstem encephalitis, acute disseminated encephalomyelitis).
- Absence of well-characterized autoantibodies in serum and CSF, and at least two of the following criteria:
- MRI abnormalities suggestive of autoimmune encephalitis[*]
- CSF pleocytosis, CSF-specific oligoclonal bands or elevated CSF IgG index, or both[*]
- Brain biopsy showing inflammatory infiltrates and excluding other disorders (e.g., tumor)
- Reasonable exclusion of alternative causes.
[] Some inherited mitochondrial and metabolic disorders can present with symmetric or asymmetric MRI abnormalities and CSF inflammatory changes resembling an acquired autoimmune disorder.102*
Figure 1. Algorithm for the diagnosis of autoimmune encephalitis.
Other poorly defined syndromes lacking established autoantibodies can be categorized as probable autoimmune encephalitis if they meet the criteria in Panel 7. When applying these criteria, consider the following points:
- CSF Findings: The absence of CSF pleocytosis does not exclude autoimmune encephalitis. For example, a significant proportion of patients with LGI1 antibody-associated encephalitis lack CSF pleocytosis. Furthermore, normal routine CSF studies do not rule out intrathecal IgG synthesis or the presence of CSF antibodies. In fact, most antibody-associated autoimmune encephalitis disorders demonstrate detectable antibodies in the CSF.
- MRI Findings: Autoimmune encephalitis can occur with normal or atypical MRI findings. Therefore, normal MRI should not automatically exclude the diagnosis in clinically suggestive cases.
- Pediatric Considerations: In children, several genetic disorders, mitochondrial diseases, and leukodystrophies can mimic autoimmune encephalitis, presenting with similar MRI and CSF abnormalities (e.g., symmetric brain involvement, pleocytosis) and even exhibiting steroid responsiveness. Careful consideration of these alternative diagnoses is crucial in pediatric cases.
For patients meeting criteria for probable autoimmune encephalitis but lacking well-characterized autoantibodies (Panel 7), further investigation for novel antibodies in specialized reference laboratories is essential. Detecting CSF antibodies reacting with the cell surface of neurons, even if the specific antigen is unknown, strongly supports the diagnosis of autoimmune encephalitis. The clinical significance of antibodies detected solely in serum, without CSF confirmation, is less clear. For example, serum GABAA receptor antibodies have been associated with a wide range of symptoms, some of uncertain clinical relevance. These advanced antibody studies are of paramount importance and may outweigh the diagnostic value of brain biopsy findings, which, while suggestive of inflammation, do not definitively establish an autoimmune cause.
In patients who do not meet criteria for probable autoimmune encephalitis and lack any autoantibodies (well-characterized or novel neuronal cell-surface antibodies), or who do not fulfill criteria for any of the aforementioned diseases and syndromes, the likelihood of an autoimmune etiology diminishes, and alternative diagnoses should be thoroughly reconsidered.
Several other autoimmune CNS disorders (e.g., primary CNS angiitis, Rasmussen’s encephalitis, Morvan’s syndrome) and diseases of unclear etiology (e.g., FIRES – febrile infection-related epilepsy syndrome) are often part of the differential diagnosis of autoimmune encephalitis (Panel 1). These conditions are discussed in detail in the appendix, emphasizing the clinical features that aid in differentiating them from autoimmune encephalitis.
Implications and Future Directions
These guidelines demonstrate the feasibility of a logical, clinically driven differential diagnosis of autoimmune encephalitis using conventional neurological assessment and standard diagnostic tests (MRI, EEG, CSF analysis). This approach allows for early establishment of probable or definite AE diagnoses and timely initiation of immunotherapy. Antibody test results, when available, can then refine the diagnosis and guide subsequent treatment strategies. Treatment recommendations for specific AE subtypes are beyond the scope of these guidelines, and evidence-based treatment algorithms remain limited for many of these disorders. A stepwise immunotherapy escalation strategy, starting with first-line therapies (steroids, IVIg, plasma exchange, or combinations) followed by second-line therapies (rituximab, cyclophosphamide, or others) for non-responders, is commonly employed, particularly in anti-NMDA receptor encephalitis and other autoimmune encephalitis types. However, rituximab is increasingly being considered as a first-line agent in certain contexts. It is important to recognize that not all autoimmune encephalitis syndromes require identical therapeutic approaches. For example, patients with LGI1 antibody-associated limbic encephalitis appear to respond more rapidly and favorably to steroids compared to those with anti-NMDA receptor encephalitis, although long-term outcomes may be better in anti-NMDA receptor encephalitis.
Future research is crucial to further refine the diagnosis and management of autoimmune encephalitis. The spectrum of AE in children differs from adults, and diagnosis in younger children is particularly challenging, suggesting that pediatric AE guidelines may rely more heavily on antibody and ancillary tests than the syndrome-based approach outlined here. Conversely, diagnosing AE in the elderly (>65 years) presents unique challenges due to the higher prevalence of age-related brain changes and comorbidities that can mimic or confound AE presentations. Future advancements will stem from cumulative clinical experience, improved differential diagnosis with AE mimics, and enhanced accessibility to rapid and reliable antibody testing, while always considering the caveats in interpretation. Continued research efforts are essential to improve our understanding, diagnosis, and treatment of autoimmune encephalitis across all age groups.
Supplementary Material
Appendix
NIHMS820429-supplement-Appendix.pdf (256.7KB, pdf)
Search Strategy and Selection Criteria.
Relevant papers were identified through PubMed searches of articles published in English up to Nov 23, 2015, using the search terms (alone or in combination): “autoimmune encephalitis”, “limbic encephalitis”, “anti-NMDA receptor encephalitis”, “ acute disseminated encephalomyelitis”, “brainstem encephalitis”, “basal ganglia encephalitis”, “Hashimoto encephalopathy”, “Rasmussen encephalitis”, “primary CNS angiitis”, “primary CNS vaculitis”, “Susac syndrome”, “Morvan syndrome”, and “neuronal autoantibodies”. Additional studies were identified from the authors’ files. The final reference list was generated on the basis of relevance to the topics covered in this Position Paper.
Acknowledgments
We thank the Autoimmune Encephalitis Alliance (USA), the Encephalitis Society (UK), the Anti-NMDA Receptor Encephalitis Foundation Inc (Canada), and the Anti-NMDA Receptor Encephalitis Patient Initiative (Germany) for disseminating information, helping patients and families, and promoting research in autoimmune encephalitis. FG was supported in part by grant 20141830 Fundació la Marató TV3. MJT has been supported by an Erasmus fellowship, the Netherlands Organisation for Scientific Research (Veni-incentive), and a grant from the Dutch Epilepsy Foundations (NEF project 14–19). RCD has received research funding from the National Health and Medical Research Council, MS Research Australia, the Tourette Syndrome Association, the University of Sydney, and the Petre Foundation. MG receives grants from the National Institute on Aging; has received grants from CurePSP and the Tau Consortium; and has received speaker’s fees and research funding from Grand Round Lectures, and the Michael J Homer Family Fund. PW is supported by the National Health Service National Specialised Commissioning Group for Neuromyelitis Optica, UK, and the National Institute for Health Research Oxford Biomedical Research Centre, and has received travel grants from the Guthy-Jackson Charitable Foundation. JD was supported by the Instituto Carlos III (FIS 14/00203) grant, National Institutes of Health RO1NS077851 grant, and Fundació Cellex.
Footnotes
[*]Altered mental status defined as decreased or altered level of consciousness, lethargy, or personality change.
[†]Brain MRI hyperintense signal on T2-weighted fluid-attenuated inversion recovery sequences highly restricted to one or both medial temporal lobes (limbic encephalitis), or in multifocal areas involving grey matter, white matter, or both compatible with demyelination or inflammation.
[*]If one of the first three criteria is not met, a diagnosis of definite limbic encephalitis can be made only with the detection of antibodies against cell-surface, synaptic, or onconeural proteins.
[†]18 Fluorodeoxyglucose (18F-FDG) PET can be used to fulfil this criterion. Results from studies from the past 5 years suggest that 18F-FDG-PET imaging might be more sensitive than MRI to show an increase in FDG uptake in normal-appearing medial temporal lobes.44,45
[*]Patients with a history of herpes simplex virus encephalitis in the previous weeks might have relapsing immune-mediated neurological symptoms (post-herpes simplex virus encephalitis).
[†]Antibody testing should include testing of CSF. If only serum is available, confirmatory tests should be included (e.g., live neurons or tissue immunohistochemistry, in addition to cell-based assay).
[*]There is no disease-specific cutoff value for these antibodies (detectable in 13% of healthy individuals).100
[*]Some inherited mitochondrial and metabolic disorders can present with symmetric or asymmetric MRI abnormalities and CSF inflammatory changes resembling an acquired autoimmune disorder.102
Contributors
FG and JD developed the idea for the Position Paper, chaired the project, wrote the initial draft of the manuscript, which was fully reviewed by MRR and MJT, and revised the manuscript. All other authors reviewed and commented on two subsequent drafts, and the complete manuscript was commented on, revised, and approved by all authors.
Declaration of interests
FG receives royalties from licensing fees to Euroimmun for the use of IgLON5 as a diagnostic test. MJT has received research funding for consultancy work for MedImmune, and a travel grant for Sun Pharma. CGB has given scientific advice to Eisai and UCB; undertaken industry-funded travel with support from Eisai, UCB, Desitin, and Grifols; obtained honoraria for speaking engagements from Eisai, UCB, Desitin, Diamed, Fresenius Medical Care; and received research support from Astellas Pharma, Octapharma, Diamed, and Fresenius Medical Care. CGB is an employee of Krankenhaus Mara, Bielefeld, Germany, which runs a laboratory for the detection of autoantibodies including those described in this paper; external senders are charged for antibody diagnostics. RCD has received research funding from the Star Scientific Foundation and Pfizer Neuroscience and speaker’s honoraria from Biogen Idec and Bristol-Myers Squibb. JMG has received compensation for medical legal consulting and for consulting on a scientific advisory board for Medimmune and Roche; he has received research funding through the University of California, San Francisco, USA, from Quest Diagnostic for work on a dementia care pathway. MG receives grants from Quest Diagnostics and has received personal fees for consultancy work from MedaCorp, Gerson-Lehman Group, Best Doctors, Advance Medical, Inc, and Optio LLC. JH receives royalties from licensing fees to Athena Diagnostics, Euroimmun, and ravo Diagnostika for a patent for the use of CV2/CRMP5 as diagnostic tests. SRI receives royalties from licensing fees to Euroimmun for patents for the use of LGI1, CASPR2, and contactin-2 as autoantibody tests. EL has received speaker’s honoraria and consultancy fees from Grifols, and consultancy fees from Medimmune. FL has received speaker’s honoraria from Grifols, Teva, and Biogen Idec and is employed by University Medical Center Schleswig-Holstein, Kiel, Germany, which offers commercial antibody testing without any personal reimbursements. MR reports that his employers, the University Hospital and Medical University of Innsbruck, Austria, receive payments for antibody assays (NMDA receptor, AQP4, and other autoantibodies) and for AQP4 antibody validation experiments organised by Euroimmun. MRR receives royalties from licensing fees to Euroimmun for a patent for the use of NMDA receptor as an autoantibody test, and from licensing fees to Athena Diagnostics for a patent for the use of Ma2. AS has received compensation for consulting services and speaker honoraria from Bayer-Schering, Merck-Serono, Biogen Idec, Sanofi-Aventis, Teva, and Novartis. AVe reports personal fees from Medimmune. AVi receives royalties from licensing fees to Euroimmun for the use of LGI1 and CASPR2 as diagnostic tests. PW receives royalties for the use of LGI1 and CASPR2 as autoantibody diagnostic tests; is a named inventor on a patent for the use of GABAA receptor as an autoantibody test; and has received speaker honoraria from Biogen Idec and Euroimmun. JD receives royalties from licensing fees to Athena Diagnostics for a patent for the use of Ma2 as an autoantibody test; licensing fees to Euroimmun for patents for the use of NMDA receptor and GABAB receptor as autoantibody tests; licensing fees for the use of DPPX, GABAA receptor, and IgLON5 antibodies as diagnostic tests; and has received a research grant from Euroimmun. RB, SB, TC, IC, CAG, RH, TI, HP, AR-G, KR, and K-PW declare no competing interests. None of the funding sources had any influence in the preparation of this Position Paper.
References
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
NIHMS820429-supplement-Appendix.pdf (256.7KB, pdf)