Introduction
Congenital Adrenal Hyperplasia (CAH) is a group of autosomal recessive genetic disorders affecting the adrenal glands. The most common cause of CAH stems from a deficiency in the enzyme 21-hydroxylase, crucial for the synthesis of cortisol and aldosterone. This deficiency, primarily due to mutations in the CYP21A2 gene, leads to an overproduction of androgens, causing virilization in females and potentially life-threatening salt-wasting crises in both sexes. Early and accurate Congenital Adrenal Hyperplasia Prenatal Diagnosis is paramount, particularly for female fetuses, where androgen excess can result in ambiguous genitalia at birth. Traditional invasive prenatal diagnostic methods, such as chorionic villus sampling and amniocentesis, pose risks and are typically performed too late in gestation to guide the earliest interventions. However, the advent of non-invasive methods, specifically Massively Parallel Sequencing (MPS) of cell-free fetal DNA in maternal plasma, offers a transformative solution for early and safe congenital adrenal hyperplasia prenatal diagnosis, potentially changing the landscape of prenatal care for families at risk.
Understanding Congenital Adrenal Hyperplasia (CAH)
Congenital Adrenal Hyperplasia (CAH) is primarily caused by mutations in the CYP21A2 gene, which provides instructions for making the 21-hydroxylase enzyme. This enzyme is essential for the adrenal glands to produce cortisol and aldosterone, hormones vital for regulating stress response, blood pressure, and electrolyte balance. When 21-hydroxylase is deficient, the body cannot produce enough cortisol and aldosterone. In response, the pituitary gland releases more adrenocorticotropic hormone (ACTH) to stimulate the adrenal glands. This overstimulation leads to the adrenal glands producing excess androgens, male sex hormones, which cause a range of symptoms depending on the severity of the deficiency and the sex of the individual.
CAH presents with a spectrum of clinical phenotypes, broadly categorized into:
- Salt-wasting CAH: The most severe form, characterized by a near-complete deficiency of 21-hydroxylase. Infants with salt-wasting CAH cannot retain sodium, leading to dehydration, low blood pressure, and potentially fatal adrenal crises shortly after birth.
- Simple virilizing CAH: A less severe form, with partial 21-hydroxylase deficiency. While cortisol production is reduced, it’s often sufficient to prevent salt-wasting crises. However, excess androgen production leads to virilization in females, including ambiguous genitalia at birth. In males, it can cause early puberty.
- Nonclassical CAH (NCCAH): The mildest form, often diagnosed later in childhood or adulthood. Individuals with NCCAH have sufficient cortisol and aldosterone production but may experience milder androgen excess symptoms like acne, hirsutism (excess hair growth), and irregular menstrual cycles in females, and early puberty in males.
For female fetuses with classical CAH, excess androgen exposure in utero, starting as early as the 9th week of gestation when genital organogenesis begins, leads to virilization of the external genitalia. This can result in ambiguous genitalia at birth, requiring surgical correction and significant psychological distress for families. Therefore, early congenital adrenal hyperplasia prenatal diagnosis is critical to facilitate timely interventions, particularly prenatal dexamethasone treatment, aimed at reducing androgen levels and mitigating virilization in affected female fetuses.
Challenges of Current Prenatal Diagnostic Methods
Traditionally, prenatal diagnosis of CAH relies on invasive procedures: chorionic villus sampling (CVS) and amniocentesis. CVS, typically performed around 11-14 weeks of gestation, involves taking a sample of placental tissue. Amniocentesis, usually conducted between 15-20 weeks, involves extracting amniotic fluid surrounding the fetus. Both procedures carry a small but significant risk of miscarriage and other complications for both mother and fetus.
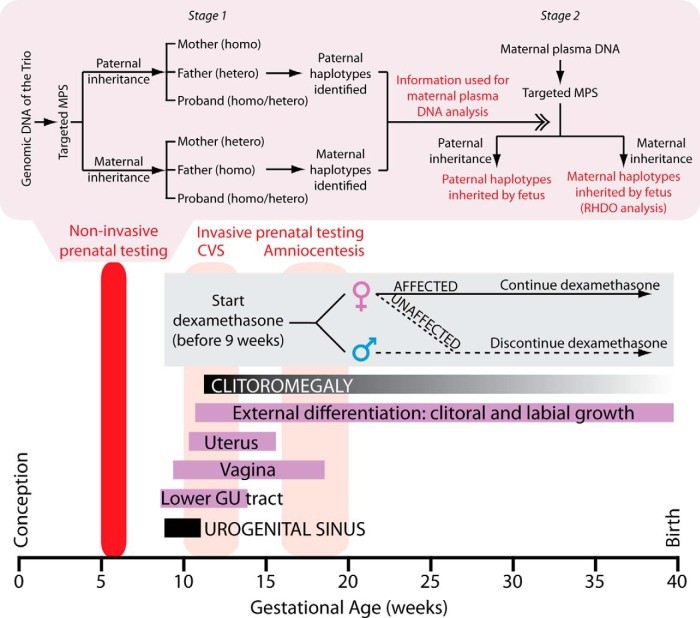
Figure 1. Temporal relationship between conventional prenatal management and targeted MPS for noninvasive detection of CAH. This diagram illustrates the timeline of genital organ development, conventional invasive diagnostic methods (CVS, amniocentesis), and the critical window for initiating dexamethasone therapy. Targeted MPS aims to provide earlier diagnosis before 9 weeks of gestation.
Furthermore, these invasive methods provide genetic results no earlier than approximately 14 weeks of gestation, well after genital organogenesis has commenced. This late diagnosis poses a significant challenge, particularly in the context of prenatal dexamethasone treatment. Dexamethasone, a synthetic glucocorticoid, can suppress fetal adrenal androgen production and prevent virilization in affected female fetuses. However, to be effective, dexamethasone treatment must begin before the 9th week of gestation. Due to the limitations of invasive prenatal diagnosis, when mothers at risk of having a CAH-affected child opt for prenatal dexamethasone treatment, it must be initiated empirically, before knowing the fetal genotype.
This empirical approach leads to a significant ethical and medical dilemma. As CAH is an autosomal recessive disorder, for each pregnancy in at-risk couples, there is only a 1 in 8 chance of having an affected female fetus requiring treatment. Consequently, in the traditional scenario, seven out of eight fetuses – males and unaffected females – are unnecessarily exposed to dexamethasone. Prenatal dexamethasone treatment is not without potential side effects for the mother and the developing fetus, including maternal weight gain, mood changes, and potential long-term effects on the child. The Endocrine Society guidelines emphasize that prenatal dexamethasone treatment should be considered experimental and performed under strict research protocols due to these concerns.
Therefore, the critical need for earlier congenital adrenal hyperplasia prenatal diagnosis is evident. A non-invasive method capable of providing accurate genetic diagnosis before the 9th week of gestation would allow for targeted dexamethasone treatment, benefiting only affected female fetuses and avoiding unnecessary exposure for the majority of pregnancies at risk.
Non-Invasive Prenatal Diagnosis using Massively Parallel Sequencing (MPS)
Massively Parallel Sequencing (MPS) of cell-free fetal DNA (cffDNA) extracted from maternal plasma has emerged as a revolutionary approach in prenatal genetics. During pregnancy, a small fraction of fetal DNA circulates freely in the maternal bloodstream. By analyzing this cffDNA, it is possible to obtain genetic information about the fetus non-invasively, simply through a maternal blood draw. However, cffDNA exists in a highly fragmented state and is present in maternal plasma amidst a vast excess of maternal DNA, making simple PCR-based analyses challenging, especially for disorders like CAH where the gene of interest (CYP21A2) has a highly homologous pseudogene (CYP21A1P).
To overcome these challenges, a targeted MPS approach combined with haplotype analysis has been developed for non-invasive congenital adrenal hyperplasia prenatal diagnosis. This strategy focuses on capturing and sequencing a specific genomic region flanking and including the CYP21A2 gene. The key steps of this approach are:
-
Trio Sequencing and Haplotype Mapping: Genomic DNA samples from the parents and a previously affected child (proband) are analyzed using targeted MPS. This allows for the identification of single nucleotide polymorphisms (SNPs) linked to the CYP21A2 gene and the construction of parental haplotypes. Haplotypes are sets of genetic variants on the same chromosome that tend to be inherited together. By analyzing the proband’s DNA, researchers can determine which parental haplotypes carry the disease-causing mutations.
-
Maternal Plasma DNA MPS and Relative Haplotype Dosage Analysis (RHDO): Cell-free fetal DNA is extracted from a maternal plasma sample, ideally obtained as early as 6 weeks of gestation. Targeted MPS is performed on the plasma DNA to analyze the representation of parental haplotypes. Relative Haplotype Dosage Analysis (RHDO) is then employed to determine the fetal inheritance of parental haplotypes. RHDO leverages informative SNPs, which are homozygous in one parent and heterozygous in the other, to track the relative amounts of each parental haplotype in the maternal plasma. An overrepresentation of a specific parental haplotype indicates fetal inheritance of that haplotype.
-
Fetal CAH Status Deduction: By determining the fetal inheritance of parental haplotypes, particularly those linked to CYP21A2 mutations, the fetal CAH status can be accurately deduced. If the fetus inherits two mutated haplotypes (one from each parent), it is diagnosed with CAH. If it inherits one mutated haplotype, it is a carrier. If it inherits no mutated haplotypes, it is unaffected.
This non-invasive MPS approach offers several significant advantages:
- Early Diagnosis: Diagnosis can be achieved as early as 5-6 weeks of gestation, well before the critical 9-week window for genital organogenesis and dexamethasone treatment initiation.
- Non-Invasive Nature: It eliminates the risks associated with invasive procedures like CVS and amniocentesis, ensuring the safety of both mother and fetus.
- Targeted Treatment: It allows for targeted dexamethasone treatment, only for mothers carrying affected female fetuses, minimizing unnecessary drug exposure for unaffected fetuses.
- Broad Applicability: This strategy is not limited to CAH and can be adapted for non-invasive prenatal testing of other autosomal recessive disorders.
Study and Results: Validation of Non-Invasive MPS for CAH
A study was conducted to validate the effectiveness of this targeted MPS approach for non-invasive congenital adrenal hyperplasia prenatal diagnosis. Fourteen families, each with a child affected by classical CAH, were recruited. The study involved trio sequencing of parents and proband to establish parental haplotypes and targeted MPS of maternal plasma DNA samples obtained as early as 5 weeks 6 days of gestation.
Figure 2. Pedigree for 14 families with CAH undergoing massively parallel sequencing and haplotype analysis. This figure illustrates the family structures, genotypes, ethnicities, Prader scores for affected females, and dexamethasone treatment history for the studied families.
[
The results demonstrated remarkable accuracy: in all 14 families, the fetal CAH status deduced by targeted MPS of maternal plasma DNA was 100% concordant with the diagnosis established by invasive prenatal testing or newborn diagnosis. This included the correct identification of seven affected fetuses, five carriers, and two unaffected fetuses. Notably, in family N, diagnosis was achieved from a maternal plasma sample collected at 5 weeks 6 days of gestation, showcasing the potential for very early congenital adrenal hyperplasia prenatal diagnosis using this method.
Figure 3. Fetal inheritance of parental mutations in family A. This figure details the haplotype analysis process for family A, illustrating paternal and maternal inheritance determination using informative SNPs and Relative Haplotype Dosage Analysis (RHDO).
For example, in Family A, where both parents were carriers of a 30-kb deletion in the CYP21A2 locus, targeted MPS of maternal plasma at 8 weeks 5 days correctly identified that the fetus inherited the mutated haplotype from both parents, indicating an affected fetus. Conversely, in Family C, the analysis correctly identified an unaffected fetus inheriting only one mutated haplotype, making it a carrier.
These findings underscore the robustness and reliability of non-invasive MPS for early congenital adrenal hyperplasia prenatal diagnosis.
Clinical Implications and Advantages of Early Non-Invasive CAH Diagnosis
The successful validation of non-invasive MPS for congenital adrenal hyperplasia prenatal diagnosis has significant clinical implications and offers numerous advantages over conventional methods.
Firstly, the ability to diagnose CAH as early as 5-6 weeks of gestation provides a crucial window for timely intervention. For affected female fetuses, this early diagnosis enables the initiation of prenatal dexamethasone treatment before the onset of significant genital virilization, potentially preventing or minimizing ambiguous genitalia at birth. This can significantly improve the quality of life for affected females and reduce the need for extensive surgical corrections and associated psychological distress.
Secondly, the non-invasive nature of the method eliminates the risks associated with CVS and amniocentesis, making prenatal diagnosis safer for both mother and fetus. This is particularly important in developing countries where access to sterile facilities for invasive procedures may be limited, and the risk of complications is potentially higher. Non-invasive prenatal diagnosis through a simple blood draw can be readily implemented in diverse healthcare settings.
Thirdly, early and accurate diagnosis allows for targeted dexamethasone treatment. By identifying unaffected fetuses and male fetuses (who do not benefit from prenatal dexamethasone for genital outcomes), unnecessary dexamethasone exposure can be avoided. This reduces potential side effects for both mothers and unaffected children and aligns with ethical considerations regarding fetal exposure to medication.
Figure 4. Fetal haplotype analysis in families A to N. This figure summarizes the haplotype analysis results for all 14 families, illustrating paternal and maternal haplotype inheritance patterns and recombination events across the CYP21A2 gene region.
Beyond CAH, the successful application of non-invasive MPS and haplotype analysis for congenital adrenal hyperplasia prenatal diagnosis paves the way for its broader application to other autosomal recessive disorders. This technology holds promise for expanding non-invasive prenatal testing menus and improving prenatal care for a wide range of genetic conditions.
Conclusion
The development and validation of targeted MPS for non-invasive congenital adrenal hyperplasia prenatal diagnosis represent a significant advancement in prenatal medicine. This approach offers a safe, accurate, and early diagnostic method, revolutionizing prenatal care for families at risk of CAH. By enabling diagnosis before the onset of genital organogenesis, it facilitates timely and targeted interventions, particularly prenatal dexamethasone treatment for affected female fetuses, while minimizing unnecessary exposure for unaffected pregnancies. This breakthrough not only improves outcomes for individuals with CAH but also sets a precedent for non-invasive prenatal diagnosis of other monogenic disorders, promising a future of safer and more effective prenatal genetic testing. Further large-scale prospective studies are warranted to fully establish the sensitivity and specificity of this method and to broaden its clinical implementation, ensuring that families worldwide can benefit from this transformative technology.
References
[1] (Reference 1 from original article)
[2] (Reference 2 from original article)
[3] (Reference 3 from original article)
[4] (Reference 4 from original article)
[5] (Reference 5 from original article)
[6] (Reference 6 from original article)
[7] (Reference 7 from original article)
[8] (Reference 8 from original article)
[9] (Reference 9 from original article)
[10] (Reference 10 from original article)
[11] (Reference 11 from original article)
[12] (Reference 12 from original article)
[13] (Reference 13 from original article)
[14] (Reference 14 from original article)
[15] (Reference 15 from original article)
[16] (Reference 16 from original article)
[17] (Reference 17 from original article)
[18] (Reference 18 from original article)
[19] (Reference 19 from original article)
[20] (Reference 20 from original article)
[21] (Reference 21 from original article)
[22] (Reference 22 from original article)
[23] (Reference 23 from original article)