Introduction
Craniosynostosis, the premature fusion of cranial sutures, leads to abnormal skull development and affects approximately 1 in 2000–2500 births for isolated cases [1]. Syndromic craniosynostosis, a more complex entity, involves multiple suture fusions alongside malformations in other organ systems [2]. Accurate prenatal diagnosis of syndromic craniosynostosis is crucial for guiding perinatal management and providing informed interdisciplinary counseling to parents. While isolated craniosynostosis is often sporadic, syndromic forms are frequently associated with genetic syndromes such as Apert, Crouzon, Pfeiffer, and Saethre-Chotzen syndromes. Among these, Apert syndrome is the most prevalent, occurring in approximately 1 in 100,000 live births [3], often stemming from de novo mutations in the Fibroblast Growth Factor Receptor 2 (FGFR2) gene [4].
Apert syndrome is characterized by bicoronal craniosynostosis, midface hypoplasia, and distinctive syndactyly of the hands and feet [5, 6]. Central nervous system abnormalities, including corpus callosum malformations, limbic system anomalies, and gyral abnormalities, are also commonly observed [7], potentially leading to neurological developmental delays ranging from mild to severe [6]. Postnatal management often involves surgical intervention to address increased intracranial pressure.
Crouzon syndrome, another autosomal dominant disorder caused by FGFR2 gene mutations [8], shares features with Apert syndrome, including bicoronal craniosynostosis and midface hypoplasia. However, Crouzon syndrome typically presents with milder malformations, and limb involvement is rare [9]. Pfeiffer syndrome, also characterized by bicoronal craniosynostosis and syndactyly, is caused by autosomal dominant mutations in FGFR1 or FGFR2 genes [10, 11]. Pfeiffer syndrome can involve additional sutures and may present with skeletal, central nervous system, and gastrointestinal anomalies [12]. Saethre-Chotzen syndrome, caused by autosomal dominant mutations in the TWIST and FGFR2 genes [13, 14], is distinguished by milder craniosynostosis affecting various cranial sutures and syndactyly.
Prenatal ultrasound plays a pivotal role in the early detection of syndromic craniosynostosis [5]. Differentiating between these syndromes and distinguishing them from non-syndromic craniosynostosis is paramount for accurate genetic counseling and tailored perinatal care [15]. Fetal Magnetic Resonance Imaging (MRI) is a valuable adjunct to confirm ultrasound findings and identify potential central nervous system malformations [5].
This study aims to elucidate the prenatal sonographic signs crucial for the differential diagnosis of syndromic craniosynostoses. By highlighting these sonographic markers and associated malformations, we seek to enhance awareness and facilitate early, precise prenatal diagnoses. This is essential for optimal perinatal management and comprehensive interdisciplinary counseling for families affected by these complex conditions.
Materials and Methods
We retrospectively reviewed cases from the Viewpoint (GE, Solingen, Germany) and SAP (Walldorf, Germany) patient databases at the Department of Obstetrics and the Department of Pediatric Surgery at Charité—Universitätsmedizin Berlin, spanning from 2000 to 2019. Our search strategy involved keywords indicative of abnormal head biometry, brain anomalies, and limb abnormalities, as well as specific syndrome names (Apert, craniosynostosis, Crouzon, Saethre-Chotzen, Pfeiffer).
This initial search yielded 389 cases with abnormal findings. Manual review refined this cohort to thirteen fetuses suspected of syndromic craniosynostosis. We further cross-referenced our findings with surgical records from the Department of Pediatric Neurosurgery to ensure case completeness. No additional cases were identified. For each case, we analyzed prenatal ultrasound findings, fetal MRI scans (when available), genetic testing results, delivery mode, and postnatal procedures.
Results
Our analysis identified thirteen pregnancies with a high prenatal suspicion of syndromic craniosynostosis between 2000 and 2019. Table 1 details the specific sonographic findings for each syndrome. Apert syndrome was suspected in ten cases based on characteristic sonographic features. Molecular genetic testing in these cases revealed a p.(Pro253Arg) mutation in the FGFR2 gene in five, confirming the diagnosis. Interestingly, one case initially suspected as Apert syndrome based on sonographic presentation was genetically diagnosed postnatally with Greig cephalopolysyndactyly syndrome due to a GLI3-gene mutation [16]. Another case revealed a p.(Pro253Leu) mutation in the FGFR2 gene. Parental testing identified the same mutation in the father, who was previously undiagnosed, highlighting the variable expressivity of these conditions. Three families declined genetic testing.
Table 1. Sonographic findings in syndromic craniosynostosis
| Syndrome | Sonographic findings |
|---|---|
| Apert syndrome (n = 8) | Syndactyly (8/8) Frontal bossing (5/8) Cloverleaf skull (4/8) Turricephaly (2/8) Dolichocephaly (1/8) Polyhydramnios (1/8) A. lusoria dextra (1/8) Mild ventriculomegaly (1/8) Dysgenesis of corpus callosum (1/8) Cleft palate (1/8) Retracted bridge of the nose (1/8) |
| Saethre Chotzen syndrome (n = 2) | Turricephaly (1/2) Saddle nose (1/2) Flat facial profile (1/2) |
| Crouzon syndrome (n = 1) | Flattened occiput Depressed frontoparietal bones Protruded bulbi Small thorax with short ribs |
| Greig cephalopolysyndactyly syndrome (n = 1) | Agenesis of corpus callosum Hypertelorism Right-sided aortic arch Polydactyly |
Two cases were diagnosed with Saethre Chotzen syndrome, and one with Crouzon syndrome. Prenatal suspicion arose between 20 + 1 and 33 + 4 weeks of gestation. Nine diagnoses were made in the second trimester, and four in the third. Fetal MRI, recommended post-2017, was performed in three cases, confirming sonographic findings.
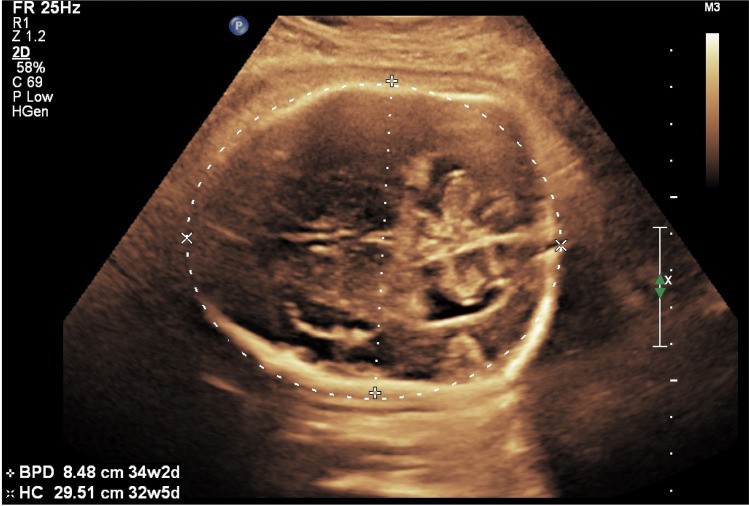
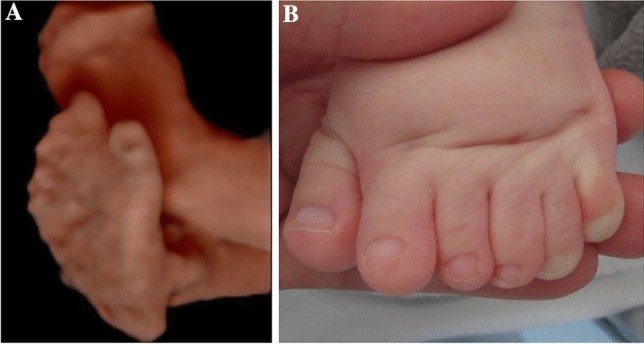
In Apert syndrome cases, key sonographic features included frontal bossing (5/8) and cloverleaf skull (4/8) (Table 1, Figs. 1a, 1b, 2a–e). Turricephaly, indicative of coronal craniosynostosis, was noted in two cases (Tables 1, 2). Prenatal ultrasound consistently detected syndactyly in all Apert syndrome cases (Fig. 1c).
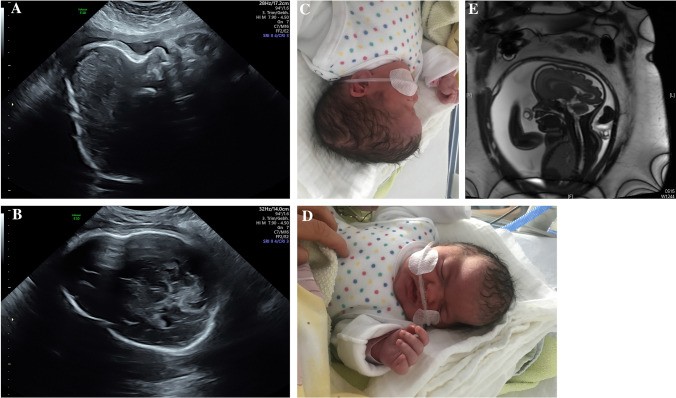
Fig. 1.
Fetus with Apert syndrome at 30 + 3 weeks gestation exhibiting frontal bossing (a, b) and bilateral syndactyly (c) as revealed by prenatal ultrasound.
Fig. 2.
A child with Apert syndrome. Prenatal ultrasound at 31 + 3 weeks gestation shows a prominent skull shape with bicoronal and sagittal craniosynostosis and frontal bossing (a, c). Prenatal MRI confirms these findings (e). Postnatal presentation includes scaphocephaly with a long, narrow skull and high forehead (d). Genetic analysis revealed a FGFR2-gene mutation (Pro253Leu), also present in the father. This mutation is located at the same amino acid position as the pathogenic p.Pro253Arg mutation associated with Apert syndrome.
In Saethre Chotzen syndrome, bicoronal craniosynostosis was prenatally diagnosed in one case, along with saddle nose and flat facial profile (Table 1). Syndactyly of hands and feet was diagnosed postnatally. In the other Saethre Chotzen case, only a prominent forehead was detected prenatally.
The Crouzon syndrome fetus presented with a flattened occiput, depressed frontoparietal bones (Fig. 3), protruding eyes (bulbi), and a small thorax with short ribs (Table 1).
Fig. 3.
Prenatal ultrasound of a fetus with Crouzon syndrome showing bilateral coronal synostosis, characterized by a flattened occiput and mild bilateral frontal depressions of the skull.
The Greig cephalopolysyndactyly syndrome fetus showed hypertelorism, agenesis of the corpus callosum, right-sided aortic arch, and polydactyly (Table 1, Figs. 4a, 5a). Postnatal examination confirmed these findings (Figs. 4b, 4c, 5b) and revealed additional malformations, including a subaortic ventricular septal defect and foot deformation.
Fig. 4.
Sonographic and postnatal images of a child with Greig cephalopolysyndactyly syndrome. Prenatal ultrasound indicated normal cranial biometric parameters but suspected dysgenesis of the corpus callosum. Postnatal images reveal a high forehead with down-slanting palpebral fissures and a low nose root.
Fig. 5.
Child with Greig cephalopolysyndactyly syndrome. Prenatal sonographic diagnosis of postaxial polysyndactyly (a) confirmed postnatally (b).
Following prenatal diagnosis and extensive interdisciplinary counseling, seven couples opted for pregnancy termination, all in cases of fetal Apert syndrome. Four of these underwent fetal autopsy, with pathoanatomical and radiological fetogram examinations corroborating sonographic findings. Among the live births, five were delivered via cesarean section, and one vaginally. Cesarean sections were performed for breech presentation (one case), preventatively (three cases), and repeat cesarean (one case).
Long-term postnatal outcome data was available for six children treated in our pediatric neurosurgery department. A child with Crouzon syndrome (born 2011) underwent decompressive craniectomy in the first year of life for increased intracranial pressure, followed by multiple craniofacial surgeries, and suffers from hearing loss and speech delay due to aural atresia. Of the two Saethre Chotzen syndrome cases, one (born 2014) had fronto-orbital remodeling at ten months, and the other (born 2019) had strip craniectomy at four months. The Apert syndrome child (born 2019) has not yet required surgery. The child with p.(Pro253Leu) mutation (born 2019) underwent biparietal strip craniectomy at two months for raised intracranial pressure. The Greig cephalopolysyndactyly syndrome child had foot surgery for hexadactyly and pes supinatus.
Discussion
Syndromic craniosynostosis, while rare, presents with characteristic prenatal sonographic features that enable diagnosis, particularly in the second trimester. Early and accurate diagnosis is vital for guiding interdisciplinary parental counseling and perinatal management. Our findings reinforce the feasibility of prenatal detection of syndromic craniosynostosis during the second trimester, aligning with our case series where the diagnosis was suspected during routine second-trimester screening in 9 out of 13 cases. The remaining 4 cases were suspected between 27 + 0 and 33 + 4 weeks of gestation. However, diagnosis can be challenging due to the variability in skull deformity severity, and standard head biometry (biparietal diameter and head circumference) may remain within normal ranges [17]. Three-dimensional ultrasonic skeletal imaging can be a valuable tool for visualizing cranial sutures and confirming suspected diagnoses (Figs. 6, 7, 8), although B-mode and conventional 3D ultrasound were sufficient for diagnosis in our cases.
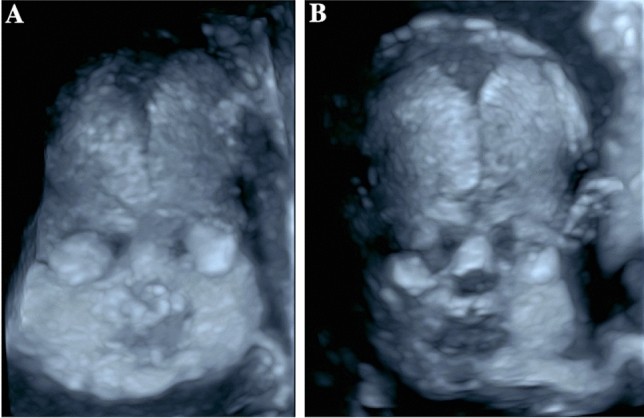
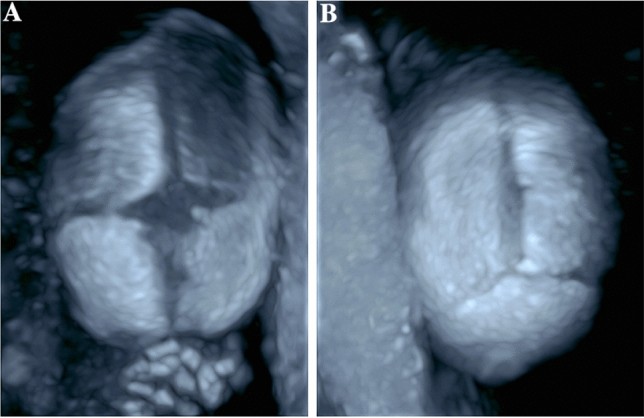
Fig. 6.
Three-dimensional skeletal ultrasound image at 27 + 5 weeks gestation showing a fetus with normal facial and skull morphology. Alt text: Normal fetal face and skull visualized with 3D skeletal ultrasound.
Fig. 7.
Three-dimensional skeletal ultrasound images of a normal fetal skull. (a) Anterior fontanelle with surrounding sutures: sutura frontalis, sutura sagittalis, and sutura coronalis. (b) Posterior fontanelle with adjacent sutures: sutura sagittalis, sutura lambdoidea. Alt text: 3D skeletal ultrasound of normal fetal skull sutures and fontanelles.
Fig. 8.
Fetus at 23 + 4 weeks gestation with partial sagittal craniosynostosis. (a) 3D skeletal ultrasound demonstrating premature fusion of the sagittal suture. (b) B-mode ultrasound indicating prominent skull shape suggestive of sagittal craniosynostosis. Alt text: Sagittal synostosis visualized by 3D and B-mode ultrasound in a fetus.
Value of Sonographic Signs in Differential Diagnosis: Skull Shapes
Abnormal skull shape emerged as the primary sonographic indicator in our series, consistent with existing literature. While basic biometric parameters may deviate from normal ranges, the specific skull shape provides critical clues for differential diagnosis [17]. The biparietal diameter (BPD) and cephalic index (CI) can be elevated or reduced depending on the affected sutures.
In our Apert syndrome cohort, all cases exhibited abnormal skull shapes (Figs. 1, 2). We observed frontal bossing (5/8), cloverleaf skull (4/8), turricephaly (2/8), and dolichocephaly (1/8) (Table 1). One Apert syndrome fetus also presented with agenesis of the corpus callosum. Midline structural anomalies, such as corpus callosum dysgenesis, are reported in up to 11% of Apert syndrome cases, along with temporal lobe alterations [7, 18].
Skull shape abnormalities were also prominent in other syndromic craniosynostoses. A Saethre-Chotzen syndrome fetus displayed turricephaly with a flat profile and saddle nose. Another presented with a small head without other skull shape anomalies. The Greig cephalopolysyndactyly syndrome fetus showed hypertelorism and agenesis of the corpus callosum. The Crouzon syndrome fetus exhibited protruding eyes.
The timing of diagnosis, predominantly in the second trimester, aligns with skull deformation development [14]. Premature suture fusion can impede visualization of underlying structures, creating a ‘brain shadowing sign,’ an early, indirect indicator of craniosynostosis, even in mild cases [19]. This sign, observable before overt skull shape changes, was reported in fetuses as early as 25 weeks gestation in a retrospective study. However, the brain shadowing sign is not specific to craniosynostosis and can also occur in fetal head molding or open spina bifida. That study also corroborated our finding of abnormal head shape as a frequent sign (23 of 24 cases), along with facial abnormalities, syndactyly, and ventriculomegaly [19]. Three-dimensional ultrasound, particularly “skeleton mode,” enhances visualization of cranial bones, sutures, and facial features compared to B-mode, aiding in detailed assessment (Figs. 6, 7, 8) [20]. Skeleton mode precisely delineates premature suture fusion (Fig. 8).
Value of Sonographic Signs in Differential Diagnosis: Associated Malformations
The presence and type of extra-cranial malformations are crucial for differentiating syndromic from isolated craniosynostosis and for distinguishing between various syndromes. Thorough sonographic examination of the hands and feet is mandatory as extremity malformations are common in syndromic cases. Limb abnormalities often provide clues to the specific syndrome. In our Apert syndrome cases, syndactyly of the upper and, in some cases, lower extremities was consistently detected prenatally (Table 1, Fig. 1c), in agreement with literature [17, 19, 20]. In one Saethre-Chotzen case, syndactyly was only diagnosed postnatally. Other reports describe postnatal findings in Saethre-Chotzen syndrome, such as anal atresia and patent ductus arteriosus [19]. Consistent with Hurst et al., we observed polydactyly in our Greig cephalopolysyndactyly syndrome case [16]. Beyond skull, central nervous system, and limb assessments, comprehensive evaluation of other organ systems is essential to identify associated malformations that can impact prognosis. Our Greig cephalopolysyndactyly syndrome case included a right aortic arch, with postnatal diagnosis of a subaortic ventricular septal defect. Congenital heart defects, including ventricular septal defects, atrial septal defects, patent ductus arteriosus, and double outlet right ventricle, are documented in Greig cephalopolysyndactyly syndrome literature [16]. Cardiac defects, alongside craniosynostosis and polydactyly, also occur in Pfeiffer syndrome and the rarer Carpenter syndrome [21]. Antley-Bixler syndrome, another rare condition, combines craniosynostosis, humero-radial synostosis, femoral curvature, joint contractures, and potential cardiac and urogenital defects [22].
Value of Fetal MRI in Differential Diagnosis
Fetal MRI, performed in three cases in our series, corroborated ultrasound findings and did not reveal additional central nervous system abnormalities (Fig. 2e). However, Rubio et al.’s study comparing ultrasound and fetal MRI in syndromic craniosynostosis identified MRI-detected dysgenesis of the corpus callosum and tethered cord syndrome not seen on ultrasound [5]. Central nervous system malformations carry neurodevelopmental implications and are critical for parental counseling. Therefore, fetal MRI should be offered in all suspected syndromic craniosynostosis cases to enhance diagnostic accuracy and inform counseling, although precise prenatal prediction of developmental outcomes remains challenging.
Value of Molecular Genetic Tests in Confirming Differential Diagnosis
Genetic testing is indispensable for differentiating isolated from syndromic craniosynostosis and confirming specific syndrome diagnoses. Parental genetic testing is important due to the possibility of autosomal dominant inheritance with variable expressivity, as well as sporadic mutations. In our study, one parent had a pre-existing Saethre-Chotzen syndrome diagnosis. In another case, genetic testing for suspected Apert syndrome revealed a p.(Pro253Leu) mutation in the FGFR2 gene in both the fetus and the previously undiagnosed father. This highlights the importance of genetic confirmation for accurate diagnosis and family counseling.
Mode of Delivery Considerations
In our case series, cesarean section was performed in five deliveries, and vaginal delivery in one. Harada et al. also reported a high cesarean section rate (73%) in fetal craniosynostosis cases [17]. In cephalic presentation without excessive head circumference increase, cesarean section is not always mandatory. However, patients should be counseled on the elevated risk of protracted labor and emergency cesarean section in cases of significant skull deformities. Delivery planning in a perinatal center is crucial to ensure optimal postnatal care, particularly regarding airway management. Prenatal consultation with a pediatric neurosurgeon is recommended to inform parents about potential surgical interventions. Postnatal cranial ultrasound and MRI, as well as echocardiography (due to increased cardiac defect prevalence), and pediatric surgical consultation are recommended.
Limitations
The rarity of syndromic craniosynostosis limited our study to thirteen cases. The retrospective design is another limitation. Direct ultrasound-MRI comparison is limited as MRI was not performed in all cases.
Conclusion
Prenatal ultrasound in the second trimester is a powerful tool for diagnosing syndromic craniosynostosis based on characteristic sonographic signs. Early and precise diagnosis facilitates targeted counseling and perinatal planning. Suspected cases warrant genetic, neonatal, and surgical workup, and fetal MRI. Given the potential for midface hypoplasia and airway compromise in newborns, delivery should be planned at a perinatal center [23]. By recognizing the key sonographic differentiators, clinicians can improve prenatal diagnosis and management of these complex conditions.
Author contribution
TC: manuscript writing, data collection, data analysis, project development. DH: manuscript editing, data analysis. WH: manuscript editing, data analysis. SV: project development, manuscript editing.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Availability of data and material
The data available on request from the corresponding author.
Code availability
Not applicable.
Declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Associated Data
Data Availability Statement
The data available on request from the corresponding author.
Not applicable.