INTRODUCTION
Cushing’s syndrome (CS), characterized by prolonged exposure to excessive cortisol, is increasingly recognized as a condition with a significant impact on adult health. While once considered rare, affecting approximately 2-3 cases per million people annually, current estimates suggest a broader prevalence, potentially ranging from 0.2% to 2% in the adult population, including subclinical presentations. This shift in understanding underscores the importance for healthcare professionals to maintain a high index of suspicion for CS to facilitate timely diagnosis and intervention. Delayed diagnosis can lead to increased morbidity and mortality, highlighting the need for effective screening and diagnostic strategies. This article aims to provide a comprehensive, stepwise Cushing’s disease diagnosis algorithm, designed to aid clinicians in navigating the complexities of hypercortisolism and ensuring accurate and efficient patient management.
While the overt manifestations of Cushing’s syndrome are often readily identifiable, the subtle or subclinical forms can be easily overlooked, particularly as they share overlapping features with common conditions such as metabolic syndrome. Conversely, it is also crucial to avoid overdiagnosis, which can lead to unnecessary investigations and patient anxiety. This review offers a structured approach to diagnosing hypercortisolism, aiming to minimize both missed and misdiagnosed cases of Cushing’s syndrome.
Step 1: Clinical Suspicion and Initial Screening for Hypercortisolism
“There is no rule more invariable than that we are paid for our suspicions by finding what we suspect.”
Henry David Thoreau
The hypothalamic-pituitary-adrenal (HPA) axis operates under a natural circadian rhythm, and disruptions to this rhythm, coupled with excessive cortisol levels, are the hallmarks of hypercortisolism. The clinical spectrum of hypercortisolism is broad, ranging from overt Cushing’s syndrome with classic signs to subclinical forms that are more challenging to detect, especially in patients presenting with obesity, diabetes, or hypertension. While features like visceral obesity, glucose intolerance, hypertension, and menstrual irregularities are common in CS, they are not specific. More discriminatory signs include skin changes such as livid striae and easy bruising (ecchymoses), muscle weakness (proximal myopathy), and skeletal issues like short stature in obese children or vertebral compression fractures. These catabolic manifestations may be subtle initially, becoming more pronounced with prolonged or severe hypercortisolism. Hirsutism on the forehead can also be a valuable clinical indicator.
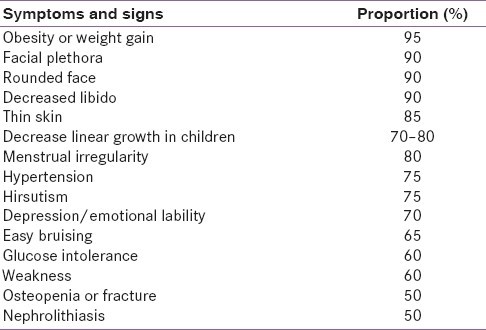
Table 1. Clinical Features of Cushing’s Syndrome
Clinical features of Cushing’s syndrome include a wide range of physical manifestations affecting various body systems.
Clinical acumen combined with a systematic approach is essential for diagnosing or excluding subclinical Cushing’s syndrome. Endocrine Society guidelines recommend screening for hypercortisolism in specific clinical scenarios:
- Patients presenting with multiple, progressive, and specific clinical findings suggestive of Cushing’s syndrome.
- Individuals with atypical presentations of common conditions, such as hypertension or osteoporosis at a young age.
- Patients with adrenal incidentalomas, discovered incidentally during imaging for other reasons.
- Children exhibiting a decline in height percentile alongside an increase in weight percentile.
Subclinical Cushing’s syndrome is observed in a notable proportion (5–30%) of adrenal incidentalomas, which themselves are found in 4–7% of the adult population. Extrapolating these figures suggests that subclinical CS prevalence in adults could range from 0.2% to 2.0%. It’s also important to consider Cushing’s syndrome in the differential diagnosis of polycystic ovary syndrome (PCOS), which is essentially a diagnosis of exclusion.
Routine screening for Cushing’s syndrome in all diabetic patients is not currently recommended, despite the reported prevalence of occult CS ranging from 1–10% in some studies. This is largely due to the high rate of false-positive results in this population. For instance, a study involving 171 obese diabetic patients found that 31 had abnormal cortisol levels on an overnight dexamethasone suppression test. However, further testing revealed elevated 24-hour urinary free cortisol in only three of these individuals, and ultimately, only one true case of Cushing’s syndrome was identified. In settings where clinical suspicion is low and the pretest probability of CS is poor, even highly sensitive tests may yield a low posttest probability, making diagnosis challenging. Therefore, widespread screening in patients with isolated components of metabolic syndrome is not generally advised without specific clinical indications.
Step 2: Establishing Endogenous Hypercortisolism
“When you have mastered numbers, you will in fact no longer be reading numbers, any more than you read words when reading books. You will be reading meanings.”
W. E. B. Du Bois
Before initiating hormonal testing, it is critical to rule out exogenous Cushing’s syndrome by taking a thorough history of exogenous glucocorticoid use. Particularly in some regions, the use of illicit or unregulated medications, including powdered or injectable steroids for pain relief, can lead to iatrogenic Cushing’s syndrome. Certain features, such as increased intraocular pressure, cataracts, benign intracranial hypertension, avascular necrosis of the femoral head, osteoporosis, and pancreatitis, are more frequently observed in exogenous CS compared to endogenous forms. Diagnosis of exogenous CS is often straightforward, supported by drug analysis and suppressed basal serum cortisol and plasma adrenocorticotropic hormone (ACTH) levels.
Conditions like depression, severe obesity, and chronic alcoholism can mimic Cushing’s syndrome, presenting as pseudo-Cushing’s syndrome, characterized by reversible HPA axis hyperactivity. These cases are typically milder clinically and may show cortisol suppression with dexamethasone testing, with normalization occurring upon treatment of the underlying condition. Tests like the insulin tolerance test or loperamide test are seldom necessary.
To quantify cortisol load, the 24-hour urinary free cortisol (UFC) test is valuable. For assessing the circadian rhythm of the HPA axis, late-night salivary cortisol and midnight serum cortisol tests are useful. The suppressibility of the HPA axis is evaluated using the overnight dexamethasone suppression test (ODST), the standard 2-day low-dose dexamethasone suppression test (LDDST), and the LDDST combined with a corticotropin-releasing hormone (CRH) stimulation test. Methodological details for these tests are summarized in Table 2. Initial diagnostic evaluation typically involves measuring either UFC, late-night salivary cortisol, or performing an ODST or LDDST. In patients with a high clinical suspicion of CS but inconclusive, discordant, or negative initial test results, further evaluation with either midnight serum cortisol levels or the LDDST-CRH stimulation test is recommended.
Table 2. Tests for Diagnosis of Endogenous Hypercortisolism
Various tests are used to diagnose endogenous hypercortisolism, each with specific methodologies and clinical applications.
Figure 1. Algorithm for Evaluating Patients Suspected of Having Cushing’s Syndrome
A diagnostic algorithm outlines the step-by-step approach for evaluating patients suspected of having Cushing’s Syndrome.
The rationale behind this stepwise approach is that initial screening tests have high sensitivity, effectively ruling out Cushing’s syndrome in most cases. If an initial test is positive, repeating the test and conducting further evaluations increases specificity, reducing the likelihood of misdiagnosis. In patients with classic Cushing’s syndrome features, all diagnostic tests generally perform well. The diagnostic challenge lies in differentiating subclinical Cushing’s syndrome from metabolic syndrome in patients with overlapping symptoms. While reported specificities for excluding CS in obese populations are around 96% for UFC, 90% for ODST, and 92% for salivary cortisol, even high specificity may not be sufficiently discriminatory for a rare condition like CS.
Consider a scenario with 1000 obese individuals, where only one has CS. Even with a 99% specific test, approximately 11 individuals would test positive (10 false positives and 1 true positive). Distinguishing the true CS case from false positives can be challenging even for experts. Therefore, when the pretest probability is low, clinicians should interpret test results at higher cut-off values and consider results from multiple tests to improve diagnostic accuracy. In uncertain cases, close clinical follow-up with a “wait-and-watch” approach may be the most prudent strategy.
Step 3: Determining the Source of Endogenous Hypercortisolism
“If you have never missed the diagnosis of ACTH dependent Cushing syndrome, and you have never been fooled attempting to establish its cause, you should refer your patients with suspected hypercortisolism to somebody who has.”
James Findling
Once exogenous Cushing’s syndrome is excluded and endogenous hypercortisolism is confirmed, the next step is to determine the source. Cushing’s disease (CD), caused by a pituitary adenoma secreting ACTH, is the most common cause of endogenous CS. Other causes are listed in Table 3.
Table 3. Various Causes of Hypercortisolism
Hypercortisolism can arise from various sources, categorized as ACTH-dependent and ACTH-independent causes.
ACTH Dependency versus Independency
Measuring basal plasma ACTH using a two-site immunometric assay is a key step in differentiating adrenal from pituitary or ectopic sources of Cushing’s syndrome. Suppressed morning plasma ACTH levels (<5 pg/ml) strongly suggest ACTH-independent CS, typically due to an adrenal adenoma or carcinoma. ACTH values between 5 and 15 pg/ml are considered equivocal. In cases of ACTH-dependent Cushing’s syndrome, ACTH levels are usually detectable or elevated. Specifically, a midnight plasma ACTH value >7.5 pg/ml has been found to be suggestive of Cushing’s disease.
However, it’s important to note that even with an autonomous cortisol-secreting adrenal adenoma, plasma ACTH levels may not be completely suppressed, and differentiation can be further complicated by the fact that unilateral or bilateral adrenal enlargement can occur in ACTH-dependent Cushing’s syndrome as well. In equivocal cases, a baseline CRH stimulation test can help distinguish between ACTH-dependent and independent causes. A plasma ACTH peak level >50% above baseline at 15 min or a cortisol peak level >50% above baseline at 30–45 min post-CRH administration favors ACTH-dependent Cushing’s syndrome.
ACTH-Dependent Cases: Cushing’s Disease versus Ectopic ACTH Syndrome
Clinical presentation can provide valuable clues in differentiating Cushing’s disease from ectopic ACTH syndrome. Women with milder hypercortisolism, slightly elevated plasma ACTH, and normal potassium levels are more likely to have Cushing’s disease. Conversely, men presenting with severe hypercortisolism, markedly elevated plasma ACTH, and hypokalemia are more suggestive of an occult ectopic ACTH-secreting tumor.
In ACTH-dependent endogenous hypercortisolism, the pretest probability of a pituitary origin (Cushing’s disease) is high, exceeding 80% overall and 90% in women. Therefore, magnetic resonance imaging (MRI) of the pituitary gland is the next recommended step. Emerging evidence suggests that spoiled gradient-recalled acquisition techniques may be more sensitive than conventional dynamic contrast-enhanced spin-echo MRI for detecting pituitary microadenomas. Pituitary MRI detects microadenomas in 50% to 70% of Cushing’s disease cases. If MRI results are negative or inconclusive, further diagnostic tests are necessary.
Historically, high-dose dexamethasone suppression test (HDDST), CRH stimulation test, and vasopressin stimulation test were used to differentiate Cushing’s disease from ectopic ACTH syndrome. However, none of these tests have demonstrated specificity exceeding 90%, meaning their diagnostic utility is not significantly better than the pretest probability of Cushing’s disease being of pituitary origin. Specifically, the HDDST has not shown reliable differentiation between Cushing’s disease and ectopic ACTH syndrome, and its continued routine use is not justified.
The most accurate test for differentiating Cushing’s disease from ectopic ACTH syndrome is bilateral inferior petrosal sinus sampling (IPSS) with CRH stimulation. A pituitary-to-peripheral ACTH gradient greater than 3 after CRH stimulation is highly suggestive of Cushing’s disease, with a reported specificity of 97%. False-negative IPSS results can occur due to anatomical variations in venous drainage, while false-positive results have been reported in rare cases of cyclical ectopic CS or CRH-secreting ectopic tumors. Lateralization of the pituitary adenoma using IPSS is possible in only 60–70% of patients, and the procedure carries a complication rate of approximately 2%. In some instances, the source of ectopic ACTH secretion remains occult and may only become apparent during follow-up.
ACTH-Independent Cushing’s Syndrome: Diverse Etiologies
In cases of ACTH-independent Cushing’s syndrome, a CT scan of the adrenal glands is crucial to characterize adrenal lesions as benign adenomas or malignant carcinomas based on lipid content and contrast washout characteristics. Primary pigmented nodular adrenocortical disease (PPNAD) and McCune-Albright syndrome are unique causes of Cushing’s syndrome in childhood. A paradoxical rise in cortisol after the low-dose dexamethasone suppression test is a characteristic feature of PPNAD.
ACTH-independent macronodular adrenal hyperplasia (AIMAH) is a rare familial condition where adrenal steroidogenesis is driven by non-ACTH peptides, such as vasopressin, glucose-dependent intestinal peptides, catecholamines, and luteinizing hormone. Specific testing protocols are available to identify the stimulating peptide in AIMAH. Interestingly, the identification of aberrant adrenal hormone receptors in AIMAH opens possibilities for novel, targeted pharmacological therapies as alternatives to adrenalectomy.
CONCLUSION
Diagnosing clinically overt Cushing’s syndrome and confirming endogenous hypercortisolism through a battery of tests is generally straightforward. The real challenge arises in subtle cases, where even experienced endocrinologists may struggle with test interpretation and may need to adopt a “wait-and-watch” approach. The fundamental principle remains: do not proceed to source localization until there is convincing biochemical evidence of hypercortisolism.
When determining the source of hypercortisolism, suppressed plasma ACTH levels and positive adrenal imaging typically point towards an adrenal etiology. In ACTH-dependent Cushing’s syndrome, a pituitary MRI showing a clear pituitary adenoma is highly informative. However, equivocal or negative MRI findings necessitate CRH-stimulated IPSS. Clinicians with experience in Cushing’s syndrome management will likely have encountered cases where relying solely on pretest probability without IPSS has led to diagnostic errors. Localizing an ectopic tumor remains a significant challenge.
Ultimately, while refining diagnostic approaches for complex Cushing’s syndrome cases is important, it is equally crucial to enhance awareness and promote informed screening for patients who would benefit from evaluation. This comprehensive Cushing’s disease diagnosis algorithm aims to support clinicians in navigating the complexities of diagnosing hypercortisolism and ensuring optimal patient care.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.