Introduction
Cystinuria is an inherited metabolic disorder characterized by the kidneys’ reduced ability to reabsorb cystine, an amino acid. This leads to elevated cystine concentrations in the urine, which, due to cystine’s poor solubility, results in the formation of cystine crystals and kidney stones. As the most common genetic cause of kidney stones, particularly in children, understanding cystinuria is crucial. For auto repair experts at xentrydiagnosis.store, while seemingly outside your immediate field, comprehending complex conditions like cystinuria exemplifies a commitment to knowledge and problem-solving, skills that are highly transferable and valuable in any profession. This article provides a detailed overview of cystinuria, focusing specifically on its diagnosis, to enhance your understanding of intricate systems, mirroring the complexity you navigate daily in vehicle diagnostics and repair.
Epidemiology of Cystinuria
Cystinuria is a prevalent monogenic kidney stone disease. It accounts for a significant proportion of kidney stone cases, specifically 1-2% in adults and a higher 6-8% in the pediatric population. In the United States, it’s estimated to affect 1 in 7,000 individuals, although prevalence rates vary globally, ranging from 1 in 2,500 to 1 in 100,000. These variations may reflect genetic diversity and underdiagnosis, as some individuals with cystinuria may not develop stones or undergo diagnostic testing. A large percentage of patients, approximately 75%, experience their first symptoms in childhood, with the average age of initial stone detection around 13 years. While onset age is generally similar across sexes, studies suggest boys may present with stones more frequently in the first three years of life.
Pathophysiology of Cystinuria
The underlying cause of cystinuria lies in a defect in the dibasic amino acid transporter located in the proximal tubules of the kidneys. This transporter, a heterodimer composed of two subunits (rBAT and b0,+AT), is responsible for the reabsorption of cysteine, ornithine, lysine, and arginine. In cystinuria, genetic mutations disrupt the function of this transporter, leading to impaired reabsorption of these amino acids and their subsequent excretion in the urine. While the excess excretion of ornithine, lysine, and arginine doesn’t typically cause clinical problems, cysteine is different. Cysteine can dimerize to form cystine, which is poorly soluble in water, especially at the normal pH of urine. This reduced solubility leads to cystine crystal formation and the development of kidney stones. Factors like urine pH and volume are crucial in determining cystine solubility, while dietary protein and sodium intake can influence urinary cystine excretion levels.
Genetic Basis of Cystinuria
Cystinuria is typically inherited in an autosomal recessive pattern. However, autosomal dominant inheritance with incomplete penetrance has also been observed. The condition is linked to mutations in two genes: SLC3A1, encoding the rBAT subunit, and SLC7A9, encoding the b0,+AT subunit. Cystinuria is classified into types A, B, and AB based on the affected gene. Type A is associated with SLC3A1 mutations, type B with SLC7A9 mutations, and type AB with mutations in both genes. Type A cystinuria is strictly autosomal recessive, with heterozygous carriers generally showing normal cystine excretion. The p.Met467Thr mutation is the most common SLC3A1 mutation. Type B cystinuria is usually autosomal recessive, but autosomal dominant inheritance has been reported. Notably, a significant portion of heterozygous SLC7A9 mutation carriers may have elevated urinary dibasic amino acid levels. The p.Gly105Arg mutation is the most frequent SLC7A9 mutation. Type AB cystinuria is rare, involving heterozygous mutations in both SLC3A1 and SLC7A9.
Table 1. Genetics of Cystinuria
| Mutated gene | SLC3A1 | SLC7A9 | SLC3A1 and SLC7A9 |
|---|---|---|---|
| Affected protein | rBAT | b0,+AT | rBAT and b0,+AT |
| Genotype | A | B | AB |
| Inheritance | AR | AR or AD with incomplete penetrance | Mixed heterozygosity |
| Prevalence | 45% | 53% | |
| Carrier phenotype | Normal cystine excretion | Elevated cystine excretion, rarely kidney stones | n/a (see type A or B) |
AR, autosomal recessive; AD, autosomal dominant.
Besides gene mutations, chromosomal deletions can also cause cystinuria. Hypotonia–cystinuria syndrome results from homozygous deletions affecting SLC3A1 and PREPL. This syndrome includes cystinuria along with hypotonia, mild facial abnormalities, intellectual disability, and growth hormone deficiency. More extensive deletions can lead to atypical hypotonia–cystinuria syndrome, which is more severe and involves mitochondrial dysfunction and intellectual disability due to the deletion of CAMKMT in addition to PREPL and SLC3A1. The most severe form, 2p21 deletion syndrome, also affecting PPM1B, is often fatal in early childhood.
Clinical Presentation: Recognizing the Signs of Cystinuria
The symptoms of cystinuria are consistent with those of obstructive urolithiasis. Patients may experience flank pain, loin pain, hematuria (blood in the urine, either visible or microscopic), vomiting, or fever, especially during acute stone passage. In children, symptoms can be less specific, including abdominal pain, dysuria (painful urination), recurrent urinary tract infections, or even vague symptoms like irritability, vomiting, and unexplained crying in younger children. Importantly, some individuals may have non-obstructive stones and remain asymptomatic, with stones detected incidentally during imaging for other reasons.
Cystinuria Diagnosis: Methods and Procedures
The diagnosis of cystinuria begins with clinical suspicion. Given that cystinuria is a common cause of kidney stones in children, particularly those with recurrent stones, it should be considered in any child presenting with nephrolithiasis. A family history of kidney stones or consanguinity (parents being closely related) should also raise suspicion. In adults, while cystine stones are less common than other types, cystinuria should be suspected in cases of recurrent stones, bilateral stones, large staghorn calculi, or a family history of cystinuria. Prenatal ultrasound findings, such as a hyperechoic fetal colon, can also be an early indicator, suggesting cystine precipitation in the colon due to high amniotic fluid cystine concentrations.
Diagnostic Modalities for Cystinuria
Several methods are available for confirming a diagnosis of cystinuria:
-
Stone Analysis: Analyzing the composition of any passed kidney stone is the gold standard. If the stone is composed of cystine, it confirms the diagnosis.
-
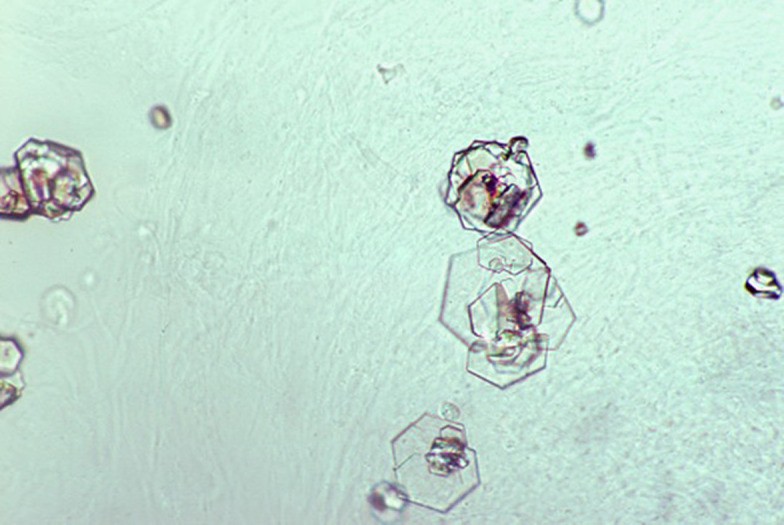
Urine Microscopy: Microscopic examination of a fresh, first morning urine sample can reveal pathognomonic hexagonal cystine crystals. These crystals are highly specific for cystinuria and can be observed in a significant proportion of untreated patients.
Characteristic hexagonal cystine crystals in urine microscopy, a key indicator for cystinuria diagnosis.
-
Qualitative Sodium Nitroprusside Test: This is a screening test where urine turns purple in the presence of high cystine levels. While useful as an initial screen, it can produce false positives in certain conditions, necessitating further confirmation.
-
Quantitative Urine Cystine Measurement: The definitive test to confirm cystine hyperexcretion is a 24-hour urine collection for quantitative chromatographic analysis of cystine. Normal daily cystine excretion is less than 300 mg/day. In children, a spot urine cystine/creatinine ratio can be used as an alternative to 24-hour collection. Age-specific normal ranges for cystine excretion are important for accurate interpretation.
-
Genetic Testing: While not always required for diagnosis, genetic testing can be valuable in atypical cases, for determining inheritance patterns, genetic counseling, and in cases of prenatal diagnosis following a hyperechoic colon finding.
Imaging in Cystinuria Diagnosis
Imaging plays a role in detecting kidney stones and assessing stone burden but is less central to the diagnosis of cystinuria itself. Cystine stones are only weakly radiopaque, making them less visible on plain X-rays compared to calcium stones. Computed tomography (CT) scans are highly sensitive for detecting cystine stones but are used judiciously in children due to radiation concerns. Renal ultrasound is often the initial imaging modality in children, with low-dose CT reserved for selected cases.
Differential Diagnosis
It is important to differentiate cystinuria from other conditions that can cause kidney stones and similar symptoms, such as:
- Other types of kidney stones: Calcium oxalate, calcium phosphate, uric acid, and struvite stones have different underlying causes and require different management strategies. Stone analysis is crucial for differentiation.
- Other causes of recurrent kidney stones: Metabolic evaluations can help rule out other conditions like hyperparathyroidism, distal renal tubular acidosis, and hyperoxaluria.
- Urinary tract infections (UTIs): While UTIs can be a symptom in cystinuria, recurrent UTIs without stone formation should prompt investigation for other causes.
- Abdominal pain of other etiologies: Especially in children, abdominal pain can be nonspecific. Cystinuria should be considered in the context of recurrent abdominal pain with other urinary symptoms or a family history of kidney stones.
Complications and Prognosis of Untreated Cystinuria
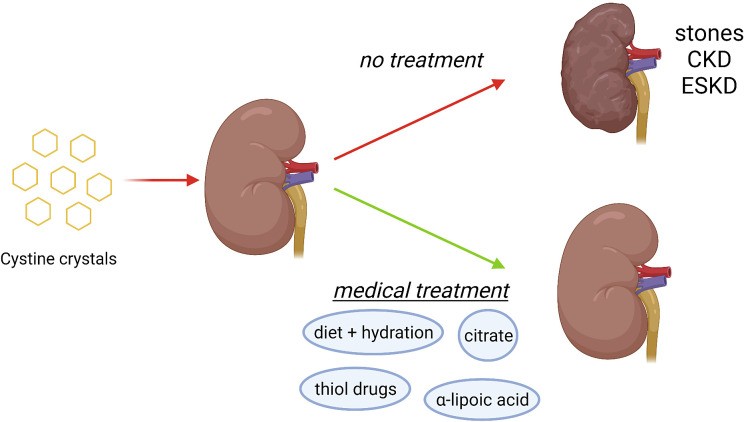
Cystinuria is associated with a significantly higher risk of chronic kidney disease (CKD) compared to other types of kidney stone formers. Studies have shown that a substantial proportion of cystinuria patients have reduced estimated glomerular filtration rate (eGFR), a marker of kidney function. End-stage kidney disease (ESKD) has also been reported in a percentage of patients. The primary mechanism of kidney damage is the chronic deposition of cystine crystals within the kidney tubules, leading to obstruction, inflammation, and fibrosis. Recurrent surgical interventions and infections can further contribute to renal injury. Hypertension is also more prevalent in cystinuria patients, likely linked to the high incidence of CKD. Therefore, early and effective diagnosis and lifelong management are essential to prevent complications and preserve renal function.
Understanding the complications of cystinuria and the importance of preventive treatment in mitigating long-term kidney damage.
Medical Management Following Cystinuria Diagnosis
Following a confirmed diagnosis of cystinuria, the focus shifts to medical management aimed at preventing stone formation and its complications. The cornerstone of treatment involves reducing urinary cystine supersaturation by increasing cystine solubility and/or decreasing urinary cystine excretion. This is achieved through a stepwise approach:
-
Dietary Modifications: Sodium restriction is generally recommended to reduce urinary cystine excretion. Reducing dietary protein intake, particularly animal protein rich in methionine (a cysteine precursor), can also be beneficial. However, in children, excessive protein restriction is avoided to ensure proper growth.
-
Hydration: Increased fluid intake is crucial to dilute urine and reduce cystine concentration. The goal is to achieve a urine volume sufficient to keep 24-hour urine cystine concentration below 250 mg/L. This often requires drinking fluids frequently throughout the day and night.
-
Urine Alkalinization: Increasing urine pH enhances cystine solubility. Potassium citrate is commonly used to raise urine pH to the target range of 7.5-8.0. Urine pH monitoring is essential to ensure effective alkalinization and to avoid the risk of calcium phosphate stone formation at excessively high pH levels.
-
Cystine-Binding Thiol Drugs: For patients who continue to form stones despite the above measures, cystine-binding thiol drugs like tiopronin, d-penicillamine, or captopril may be necessary. These drugs contain sulfhydryl groups that react with cystine to form more soluble compounds, reducing the amount of free cystine available to form stones. However, these drugs can have significant side effects, necessitating careful monitoring.
-
Emerging Therapies: Research continues to explore new treatment options for cystinuria, including crystal growth inhibitors and alpha-lipoic acid, showing promise in preclinical and early clinical studies. Vasopressin antagonists are also being investigated for their potential to increase urine volume and reduce cystine concentration.
Monitoring Treatment Effectiveness
Regular monitoring is crucial to assess treatment efficacy. 24-hour urine tests are recommended to track urine volume, cystine excretion, and pH. Cystine supersaturation and cystine capacity, when available, provide additional insights into urine cystine solubility. Treatment goals are individualized based on these parameters and clinical outcomes, aiming to minimize stone recurrence and preserve renal function.
Conclusion: The Importance of Accurate Cystinuria Diagnosis
Accurate and timely diagnosis of cystinuria is paramount for effective management and prevention of long-term complications. While cystinuria is a lifelong condition, appropriate medical treatment, guided by careful diagnosis and monitoring, can significantly reduce stone formation, preserve kidney function, and improve the quality of life for affected individuals. For professionals in fields seemingly unrelated to medicine, like auto repair experts, understanding complex diagnostic processes in conditions like cystinuria highlights the universal value of problem-solving, critical thinking, and continuous learning – skills that are essential in all fields of expertise.
Footnotes
Peer-review: Externally peer-reviewed.
Author Contributions: Concept – O.C.; Funding – O.C.; Literature Review – S.S., O.C.; Writing – S.S.; Critical Review – O.C.
Declaration of Interests: The authors have no conflict of interest to declare.
Funding: Onur Cil is supported by grants from NIH (DK126070, DK072517) and Cystic Fibrosis Foundation.