Desmoid tumors, also known as aggressive fibromatosis, are rare, benign neoplasms arising from fibrous tissue. While non-metastasizing, their locally aggressive nature and high recurrence rates pose significant clinical challenges. Accurate diagnosis is crucial for effective management, and this necessitates a thorough understanding of the differential diagnosis of desmoid tumors. This article provides an in-depth exploration of conditions that mimic desmoid tumors, ensuring clinicians can confidently differentiate and guide appropriate patient care.
Unraveling the Diagnostic Puzzle: Desmoid Tumors and Their Mimics
Diagnosing desmoid tumors can be complex due to their diverse clinical presentations and histological overlap with other conditions. The differential diagnosis is broad, encompassing both benign and malignant fibroblastic and myofibroblastic lesions. Distinguishing desmoid tumors from these mimics is paramount to avoid misdiagnosis and ensure optimal treatment strategies.
Navigating the Differential Diagnosis of Extra-Abdominal Fibromatosis
When evaluating extra-abdominal lesions, it is essential to differentiate desmoid tumors from a spectrum of conditions, ranging from benign reactive processes to malignant sarcomas.
Malignant Mimics: Fibroblastic Sarcoma and Low-Grade Fibromyxoid Sarcoma
Fibroblastic Sarcoma: This malignant tumor represents a critical differential diagnostic consideration. While both desmoid tumors and fibroblastic sarcomas can exhibit positive β-catenin staining, key histological features aid in differentiation. Fibroblastic sarcomas typically display higher cellularity, more pronounced nuclear atypia, and a characteristic herringbone growth pattern of spindle cells. Careful microscopic evaluation is crucial to identify these subtle yet significant differences.
Low-Grade Fibromyxoid Sarcoma: This sarcoma subtype can also resemble desmoid tumors. Histologically, low-grade fibromyxoid sarcoma often presents with more delicate nuclei and a distinctive network of curvilinear blood vessels. Although β-catenin staining can be positive in a subset of cases (around 30%), it is less consistent than in desmoid tumors. A definitive distinction can be achieved through fluorescence in situ hybridization (FISH) analysis to detect the characteristic FUS–CREB3L2 gene fusion, which is pathognomonic for low-grade fibromyxoid sarcoma and absent in desmoid tumors.
Benign Mimics: Gardner Fibroma, Keloids, and Reactive Fibroblastic Proliferations
Gardner Fibroma: This rare benign neoplasm occurs in individuals with familial adenomatous polyposis (FAP) and can arise in similar locations as desmoid tumors, even preceding the development of fibromatosis. Both lesions exhibit nuclear β-catenin expression, complicating differentiation based on immunohistochemistry alone. However, Gardner fibromas are typically less cellular, with a greater abundance of collagen, and the spindle cells express CD34, which is not characteristic of desmoid tumors. Clinical correlation with FAP history is also vital in considering Gardner fibroma in the differential diagnosis.
Keloids and Reactive Fibroblastic Proliferations: Following trauma, reactive fibroblastic proliferations, including keloids and hypertrophic scars, can clinically and sometimes histologically mimic desmoid tumors. These reactive lesions usually present with a more variable histological picture, often with focal hemorrhage and inflammation. Importantly, they are typically negative for β-catenin immunostaining, a valuable feature in distinguishing them from desmoid tumors. Clinical history of trauma or surgery at the site of lesion development is also a key differentiating factor.
Differentiating Intra-Abdominal Fibromatosis: A Unique Set of Challenges
The differential diagnosis of intra-abdominal desmoid tumors presents a distinct set of considerations, involving other mesenchymal tumors and reactive conditions within the abdomen.
Gastrointestinal Stromal Tumor (GIST)
GISTs are the most common mesenchymal tumors of the gastrointestinal tract and can occur in the abdomen, necessitating differentiation from desmoid tumors. While recent reports describe the coexistence of abdominal desmoids and GISTs, distinguishing them is usually straightforward. GISTs characteristically express c-KIT (CD117), CD34, and DOG1, and are virtually always β-catenin negative. Desmoid tumors, conversely, are consistently negative for c-KIT, CD34, and DOG1, and typically β-catenin positive. Furthermore, GISTs frequently harbor mutations in KIT or PDGFRA genes, which are not found in desmoid tumors. Immunohistochemistry is highly effective in differentiating these entities.
Solitary Fibrous Tumor (SFT)
SFTs are mesenchymal tumors that can occur throughout the body, including the abdomen. Some SFT variants can exhibit significant hyalinization, making them histologically challenging to distinguish from desmoid tumors, particularly in cases with limited vascularity and subtle staghorn vessel patterns. However, SFTs typically express CD34, CD99, and Bcl-2, which are not characteristic of desmoid tumors. Immunohistochemical analysis for these markers is crucial for accurate differentiation.
Inflammatory Myofibroblastic Tumor (IMT)
IMTs are another entity in the differential diagnosis of intra-abdominal desmoid tumors. IMTs are characterized by a more cellular histology, pronounced nuclear atypia, and a prominent inflammatory background. Immunohistochemically, IMTs can be positive for ALK-1 and are β-catenin negative, aiding in their distinction from desmoid tumors, which are typically ALK-1 negative and β-catenin positive.
Reactive Intra-abdominal Conditions: Sclerosing Mesenteritis and Retroperitoneal Fibrosis
Sclerosing Mesenteritis: This benign inflammatory condition of the mesentery can sometimes mimic desmoid tumors radiologically. However, sclerosing mesenteritis lacks the cellular proliferation seen in desmoid tumors and is characterized by fat necrosis, chronic inflammation, and fibrosis within the mesentery. Histopathology readily differentiates these conditions.
Retroperitoneal Fibrosis: This condition, whether idiopathic (Ormond’s disease) or secondary to medications or malignancy, can also enter the differential diagnosis. Retroperitoneal fibrosis is characterized by dense fibrosis, often with a marked lymphoplasmacytic infiltrate, which is distinct from the fibroblastic proliferation of desmoid tumors. Clinical and radiological correlation, alongside histopathological examination, helps in accurate diagnosis.
The Pivotal Role of β-catenin and Genetic Analysis in Differential Diagnosis
Nuclear β-catenin staining has become an invaluable tool in the diagnosis of desmoid tumors, observed in approximately 80% of cases. However, it is crucial to recognize that β-catenin staining is not entirely specific and can be seen in other tumors. Therefore, while positive β-catenin staining supports the diagnosis of desmoid tumor, it should be interpreted in the context of clinical and histological findings.
Genetic analysis for CTNNB1 mutations, particularly in exon 3, further enhances diagnostic accuracy. These mutations are found in approximately 85% of sporadic desmoid tumors. Detecting a CTNNB1 mutation can be particularly helpful when histological differentiation is challenging, or when clinical presentation is atypical. The location of the tumor and the specific CTNNB1 mutation subtype may also have prognostic implications, as certain mutations have been associated with higher recurrence rates.
Integrating Clinical, Histopathological, and Molecular Data for Definitive Diagnosis
The differential diagnosis of desmoid tumors requires a comprehensive approach, integrating clinical presentation, radiological findings, detailed histopathological evaluation, immunohistochemistry, and molecular analysis. No single feature is definitively diagnostic, and a combination of these parameters is essential for accurate diagnosis and appropriate patient management.
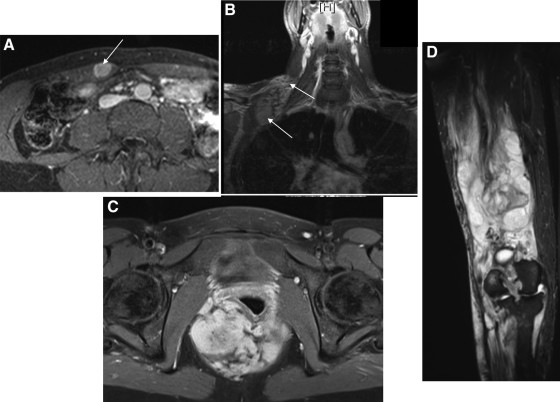
Figure 1: Typical locations of desmoid tumors, crucial for considering site-specific differential diagnoses. Understanding common locations like the rectus abdominis, head and neck, pelvis, and extremities helps narrow down differential considerations based on anatomical context.
Figure 2: Histological and immunohistochemical features aiding in differential diagnosis. (A) Classic spindle cell morphology of a desmoid tumor, a common but not specific feature requiring differentiation from sarcomas. (B) Characteristic β-catenin expression in a desmoid tumor, a helpful but not exclusive marker, as some mimics can also show positivity. Endothelial cells serve as a negative control, highlighting the specific nuclear staining in the tumor cells.
Conclusion: Precision in Diagnosis for Optimal Patient Care
The differential diagnosis of desmoid tumors is broad and complex, requiring careful consideration of various benign and malignant conditions. A detailed understanding of the clinical, histological, immunohistochemical, and molecular features of desmoid tumors and their mimics is paramount for accurate diagnosis. By integrating these diagnostic modalities, clinicians can confidently differentiate desmoid tumors from other entities, ensuring patients receive the most appropriate and effective management strategies tailored to their specific diagnosis. This precise diagnostic approach is crucial for optimizing treatment outcomes and improving the quality of life for individuals affected by desmoid tumors.
