Diabetes insipidus (DI) is a rare endocrine disorder characterized by the body’s inability to regulate fluid balance, leading to excessive urination and thirst. This condition arises from issues with antidiuretic hormone (ADH), also known as vasopressin, a crucial hormone produced by the posterior pituitary gland. ADH plays a pivotal role in signaling the kidneys to conserve water. In diabetes insipidus, either ADH is deficient, or the kidneys are unable to respond properly to it, resulting in the excretion of large volumes of dilute urine. Understanding the nuances of diabetes insipidus diagnosis is critical for effective management and improving patient outcomes.
Understanding Diabetes Insipidus
Diabetes insipidus is marked by the excretion of abnormally large volumes of dilute urine, typically exceeding 3 to 3.5 liters in 24 hours in adults, with urine osmolality less than 300 mOsmol/kg. This excessive fluid loss leads to intense thirst and a constant craving for water, particularly cold water. Unlike diabetes mellitus, which involves blood sugar regulation, diabetes insipidus centers around water balance and ADH function.
Antidiuretic hormone (ADH) is the key hormone in this condition. It is synthesized in the hypothalamus and stored in the posterior pituitary gland. ADH acts on the kidneys, specifically the distal convoluted tubules and collecting ducts, to increase water reabsorption back into the bloodstream. It achieves this by upregulating aquaporin-2 channels (AQP2) on the apical membrane surface of renal cells. These channels are essentially water pores that facilitate water movement from the nephron back into the body, concentrating the urine.
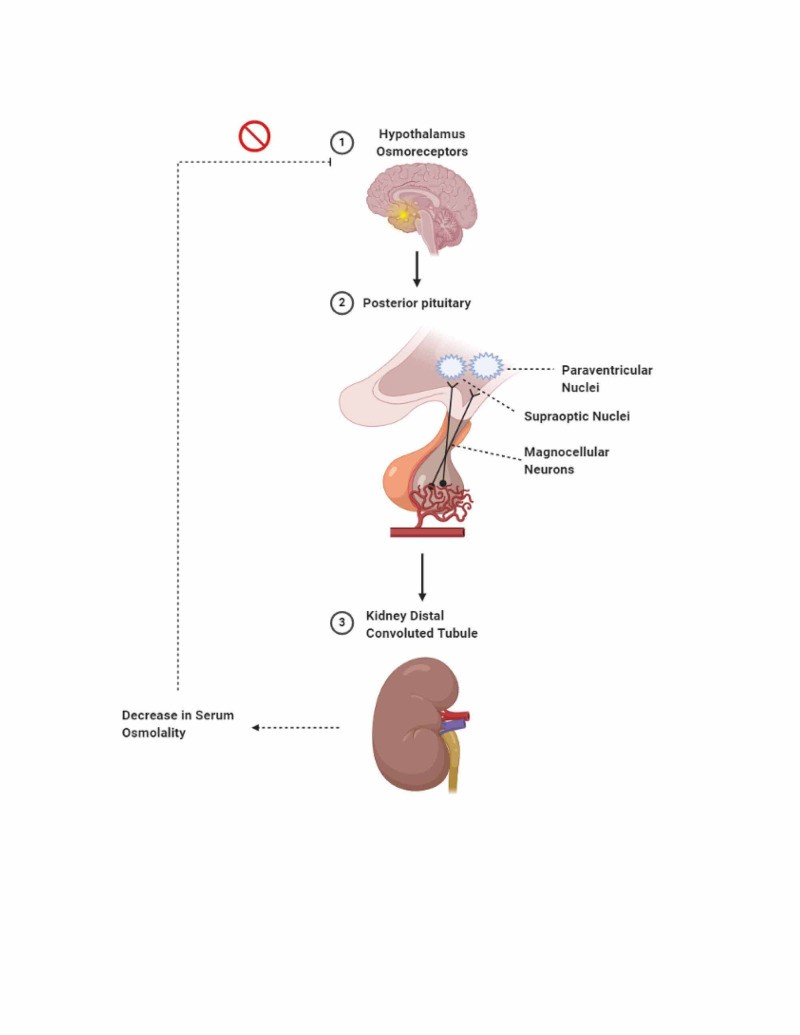
The secretion of ADH is primarily controlled by two negative feedback mechanisms: osmoregulation and baroregulation. Osmoreceptors in the hypothalamus detect even minor changes in plasma osmolality (less than 1%). An increase in osmolality triggers ADH release to conserve water. Baroreceptors respond to changes in blood volume, requiring a more significant change (5%-10% decrease) to stimulate ADH release.
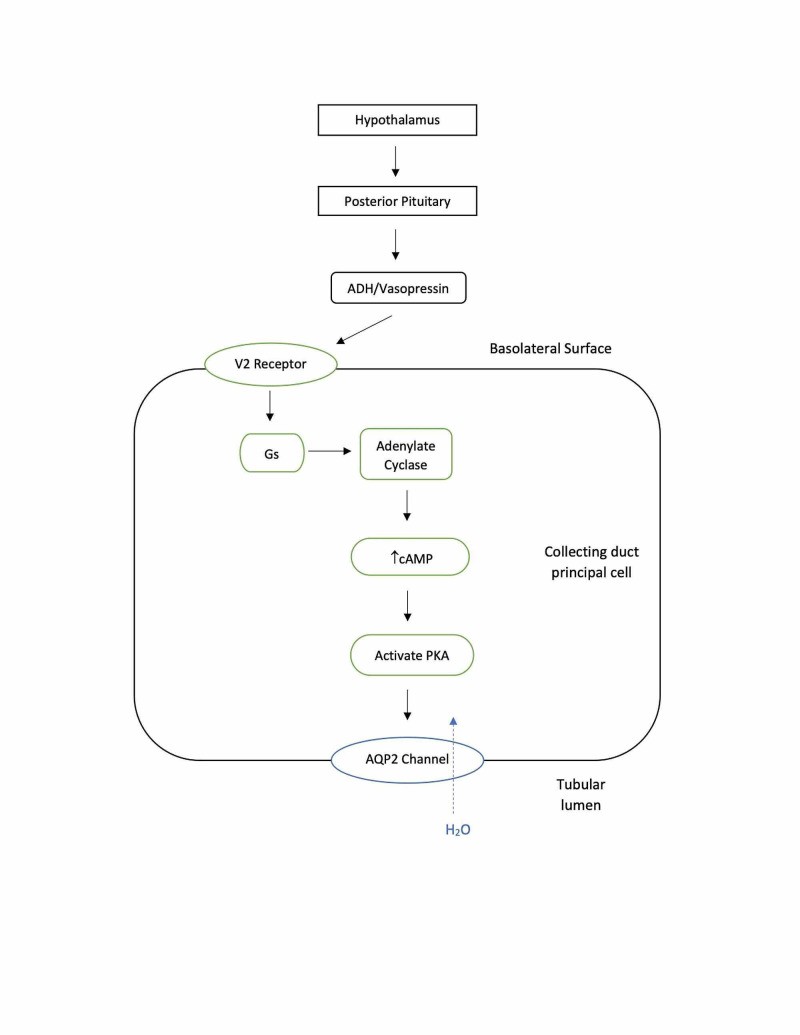
When released, ADH travels to the kidneys and binds to V2 receptors on the basolateral membrane of collecting duct cells. This binding initiates a cascade of intracellular events, increasing cyclic adenosine monophosphate (cAMP) levels, activating protein kinase A, and ultimately leading to the phosphorylation and insertion of AQP2 channels into the apical membrane. Without AQP2, the collecting duct remains largely impermeable to water, leading to water loss and dilute urine. In a healthy individual, ADH can concentrate urine up to 1200 mOsmol/kg and reduce urine output significantly. Once water balance is restored, ADH levels decrease, and AQP2 channels are downregulated. In diabetes insipidus, this intricate system malfunctions, leading to impaired water conservation.
Figure 1. Osmoregulation and ADH Release in Response to Increased Serum Osmolality
Alt text: Diagram illustrating osmoregulation: Increased serum osmolality detected by hypothalamus triggers ADH release from posterior pituitary, leading to aquaporin-2 channel insertion in kidney tubules for water reabsorption and osmolality decrease.
Figure 2. ADH Mechanism of Action in Renal Collecting Duct Cells
Alt text: Illustration of ADH function: ADH binding to receptors activates cAMP and PKA, leading to phosphorylation and insertion of AQP2 water channels into the apical membrane of renal collecting duct cells.
Types and Etiology of Diabetes Insipidus
Diabetes insipidus is categorized into four main types, each with distinct etiologies: central diabetes insipidus (CDI), nephrogenic diabetes insipidus (NDI), dipsogenic diabetes insipidus (DDI), and gestational diabetes insipidus (GDI). Accurate diagnosis requires differentiating between these types, as treatment strategies vary significantly.
Central Diabetes Insipidus (CDI)
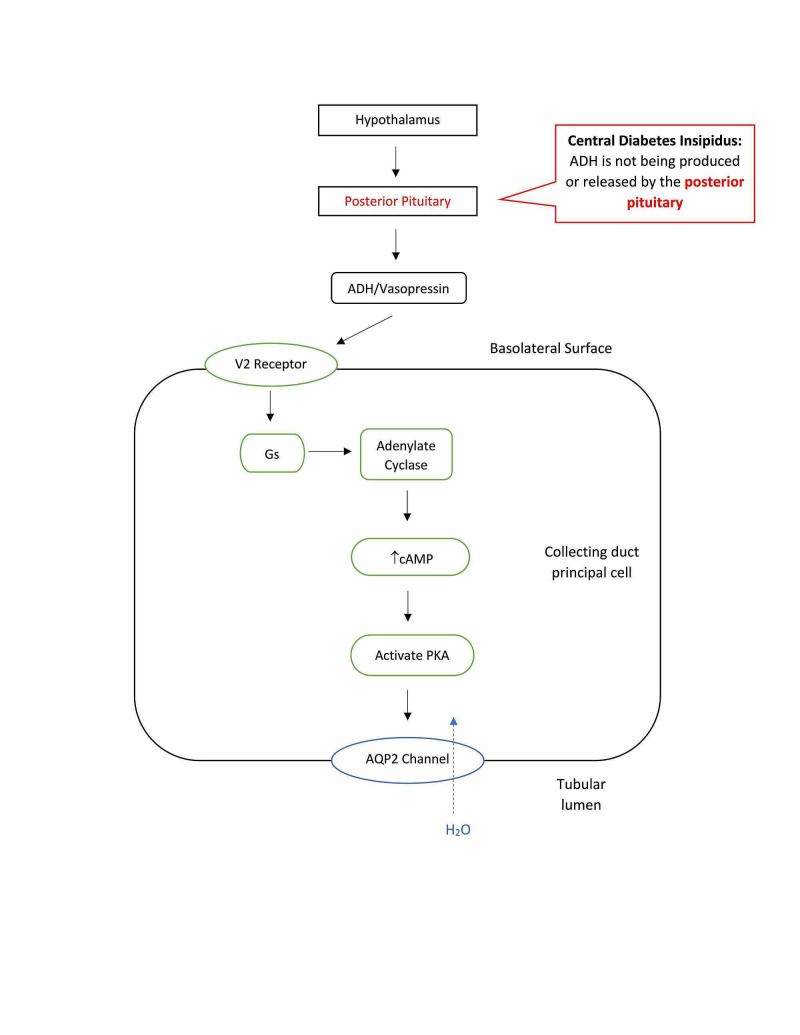
Central diabetes insipidus, also known as neurogenic DI, is the most common form. It results from a deficiency in ADH production and release from the posterior pituitary gland. This deficiency is often due to damage to the hypothalamus or pituitary gland, which can be caused by:
- Traumatic Brain Injury (TBI): Head trauma can injure the pituitary stalk, pituitary gland, or hypothalamus, accounting for a significant percentage of CDI cases.
- Neurosurgery: Iatrogenic CDI is a known complication of neurosurgery, particularly procedures involving the pituitary region.
- Tumors: Tumors in or near the hypothalamus or pituitary gland can disrupt ADH production or release.
- Infections: Infections such as meningitis or encephalitis can, in rare cases, affect the hypothalamus or pituitary gland.
- Vascular Issues: Loss of blood supply to the pituitary or hypothalamus, such as in pituitary apoplexy, can lead to CDI.
- Genetic Factors: While rare, genetic defects affecting ADH synthesis can cause familial CDI. Mutations in the AVP gene on chromosome 20p13 are most commonly implicated. Wolfram syndrome, caused by mutations in the WFS1 gene, is a rare autosomal recessive disorder that includes CDI along with diabetes mellitus, optic atrophy, and deafness.
Nephrogenic Diabetes Insipidus (NDI)
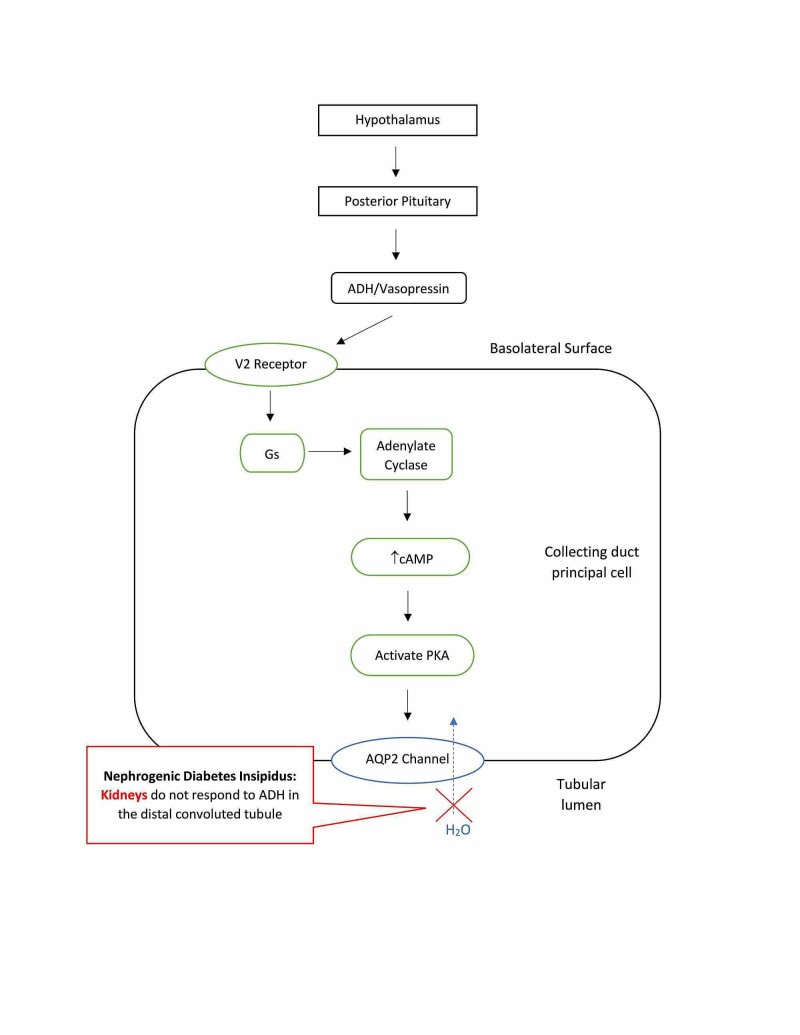
Nephrogenic diabetes insipidus occurs when the kidneys, specifically the distal convoluted tubules and collecting ducts, are resistant to ADH. In NDI, the kidneys do not respond properly to circulating ADH, leading to impaired water reabsorption. The causes of NDI are broadly classified as acquired and congenital:
- Acquired NDI: This is the most common form in adults.
- Lithium Therapy: Lithium, used to treat bipolar disorder, is a leading cause of acquired NDI. It can accumulate in renal collecting duct cells, disrupting AQP2 expression.
- Medications: Other drugs like foscarnet and clozapine, though less common, have been linked to drug-induced NDI.
- Electrolyte Imbalances: Hypercalcemia and hypokalemia can impair kidney responsiveness to ADH.
- Protein Malnutrition: Severe protein deficiency can affect renal function.
- Chronic Kidney Disease: Underlying kidney disease can reduce the kidneys’ ability to respond to ADH.
- Ureteral Obstruction Relief: Paradoxically, relief of urinary obstruction can sometimes lead to transient NDI.
- Congenital NDI: Rare genetic mutations affecting the AQP2 gene or the V2 receptor gene can cause congenital NDI. Inheritance patterns can be X-linked (most common), autosomal recessive, or autosomal dominant.
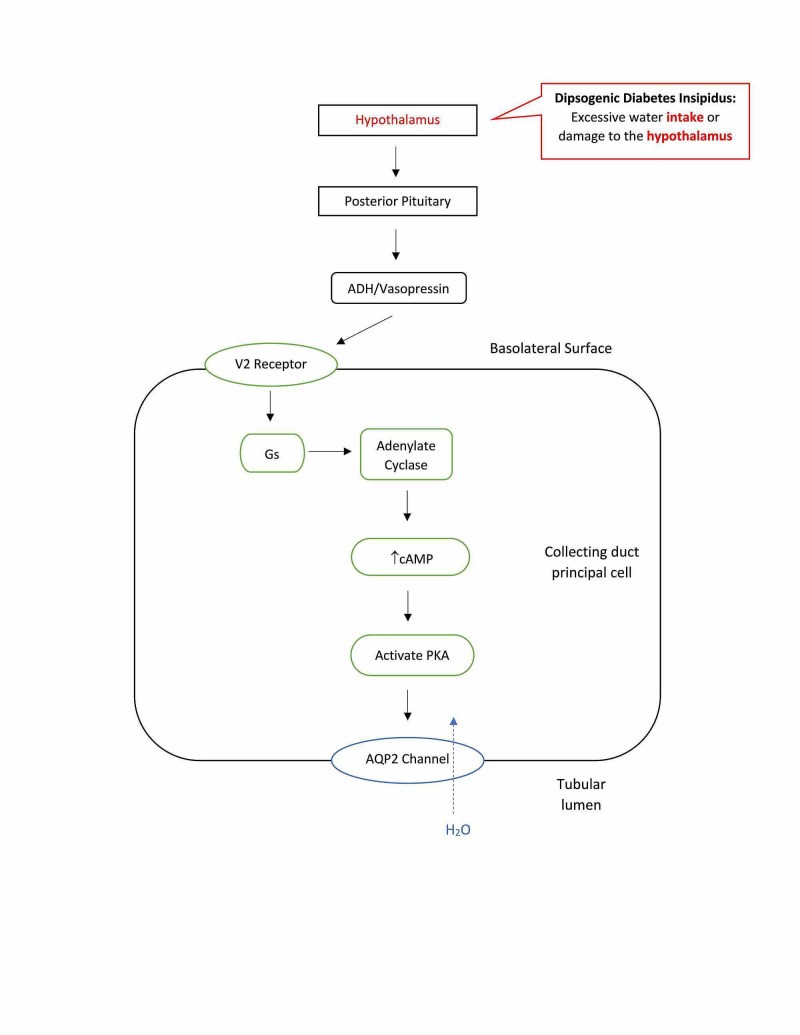
Dipsogenic Diabetes Insipidus (DDI)
Dipsogenic diabetes insipidus, also known as primary polydipsia, is characterized by excessive thirst due to a dysregulation of the thirst mechanism in the hypothalamus. In DDI, the osmotic threshold for thirst is abnormally low, leading to increased fluid intake and secondary suppression of ADH secretion. While technically not a true diabetes insipidus in terms of ADH deficiency or resistance, it presents with similar symptoms of polyuria and polydipsia. Causes can include:
- Hypothalamic Damage: Damage to the hypothalamus from injury, surgery, tumors, or infiltrative diseases.
- Psychiatric Disorders: DDI is frequently observed in individuals with psychotic or neurodevelopmental disorders.
- Brain Lesions: Lesions in brain regions involved in thirst regulation, such as the amygdala or hippocampus.
- Genetic Predisposition: Polymorphisms in the orexin 1 receptor gene have been implicated in DDI.
- Stress-Reducing Behaviors: Certain behaviors that release dopamine can trigger excessive thirst.
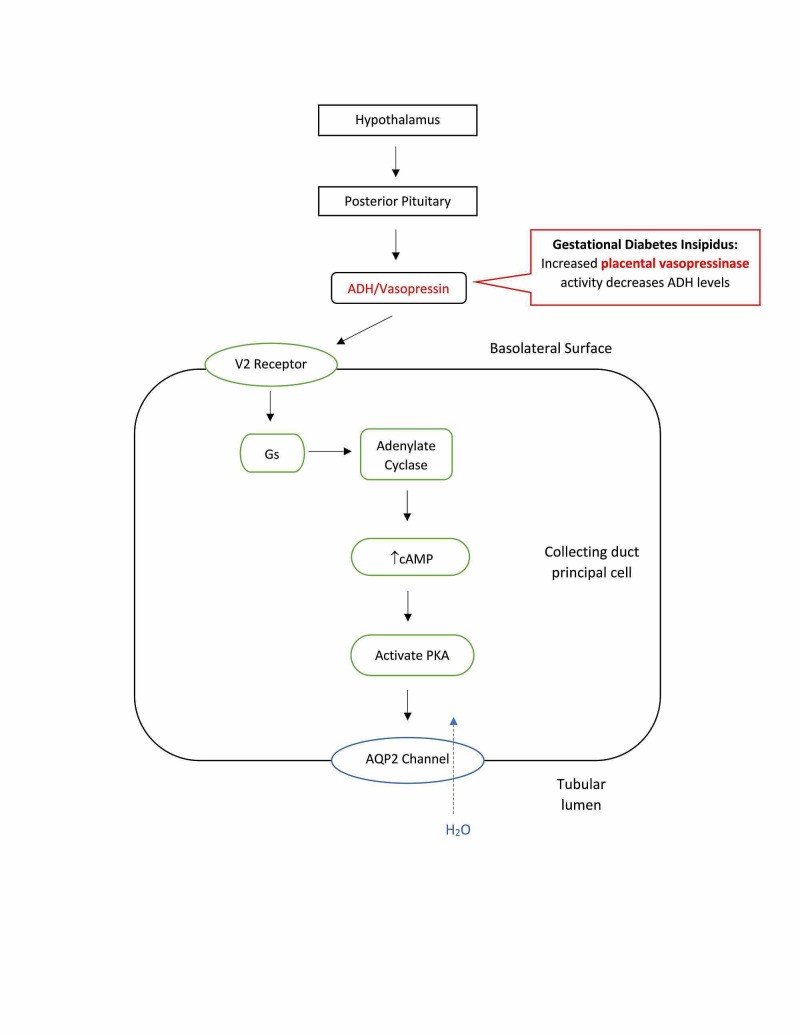
Gestational Diabetes Insipidus (GDI)
Gestational diabetes insipidus occurs specifically during pregnancy. It is caused by increased levels of placental vasopressinase, an enzyme produced by the placenta that degrades ADH. This enzymatic degradation leads to a relative ADH deficiency, resulting in polyuria. GDI typically manifests in the second or third trimester when vasopressinase levels are highest. Risk factors and contributing factors include:
- Placental Vasopressinase: Increased production of vasopressinase by the placenta is the primary cause.
- Multiple Pregnancies: Twin or multiple pregnancies are associated with higher vasopressinase levels.
- Pre-existing Asymptomatic DI: Women with mild, undiagnosed DI prior to pregnancy may become symptomatic during pregnancy due to increased ADH degradation.
- Physiological Changes of Pregnancy: Pregnancy-related hormonal changes, such as increased progesterone and corticosteroids, can also contribute to decreased ADH activity.
- Liver Dysfunction: Conditions like acute fatty liver of pregnancy or HELLP syndrome can impair liver function and reduce vasopressinase clearance, exacerbating GDI.
Figure 3. Central Diabetes Insipidus: ADH Deficiency
Alt text: Diagram of Central Diabetes Insipidus: Damage to pituitary gland leads to reduced ADH production, resulting in decreased water reabsorption and dilute urine excretion.
Figure 4. Nephrogenic Diabetes Insipidus: Renal Resistance to ADH
Alt text: Illustration of Nephrogenic Diabetes Insipidus: Renal tubules defect causes resistance to ADH, impairing water reabsorption and leading to excretion of large volumes of dilute urine.
Figure 5. Dipsogenic Diabetes Insipidus: Dysregulated Thirst Mechanism
Alt text: Visual representation of Dipsogenic Diabetes Insipidus: Excessive fluid intake due to hypothalamic thirst mechanism damage or dysfunction leads to dilute urine output.
Figure 6. Gestational Diabetes Insipidus: Placental Vasopressinase
Alt text: Diagram illustrating Gestational Diabetes Insipidus: Increased placental vasopressinase degrades maternal ADH, causing reduced ADH effect and excretion of large amounts of dilute urine.
Pathophysiology and Clinical Presentation
Regardless of the specific etiology, all forms of diabetes insipidus share a common pathophysiological pathway leading to polyuria, polydipsia, and dehydration. The disruption of ADH action or production impairs the body’s ability to conserve water, leading to significant fluid loss.
Electrolyte imbalances are a hallmark of DI. Hypernatremia, characterized by serum sodium levels above 145 mEq/L, is typical in central and nephrogenic DI, reflecting the loss of free water. Conversely, hyponatremia might be seen in primary polydipsia due to excessive water intake. Serum osmolality is also elevated in DI, typically exceeding 295 mOsm/kg, while urine osmolality is inappropriately low (below 300 mOsm/kg).
Clinical manifestations of diabetes insipidus include:
- Polyuria: Excessive urination, often with urine output exceeding 3 liters per day in adults, and even higher in severe cases.
- Polydipsia: Intense and persistent thirst, with a strong preference for cold beverages.
- Nocturia: Frequent urination at night, disrupting sleep.
- Dehydration: If fluid intake cannot keep pace with fluid loss, dehydration can occur, leading to symptoms like dry mouth, poor skin turgor, and weakness.
- Hypernatremia Symptoms: In severe cases, hypernatremia can cause neurological symptoms like confusion, irritability, lethargy, and in extreme situations, seizures and coma.
The body’s normal homeostatic mechanisms, osmoregulation and baroregulation, are overwhelmed in DI. The osmoregulatory feedback loop, normally triggered by increased serum osmolality, fails to adequately increase water reabsorption. Similarly, the baroregulatory loop, which responds to decreased blood volume, may be activated but insufficient to compensate for the profound water loss. In severe cases, baroreceptor stimulation of ADH release may take precedence over osmoregulation in an attempt to maintain blood volume.
Diabetes Insipidus Diagnosis: An Up-to-Date Approach
Diagnosing diabetes insipidus accurately is crucial for initiating appropriate treatment and preventing complications. The diagnostic process involves a combination of clinical evaluation, laboratory tests, and sometimes imaging studies. An up-to-date diagnostic approach emphasizes the use of copeptin measurements and refined water deprivation tests.
Clinical Evaluation and Initial Assessment
The diagnostic workup typically begins with a thorough medical history and physical examination. Key aspects of the clinical evaluation include:
- History of Polyuria and Polydipsia: Detailed questioning about the onset, duration, and severity of excessive urination and thirst. Quantifying fluid intake and urine output is essential.
- Nocturia: Assessing the frequency and impact of nighttime urination.
- Medical History: Inquiring about any history of head trauma, neurosurgery, tumors, medications (especially lithium), psychiatric disorders, and pregnancy (in women).
- Physical Examination: Assessing hydration status, including skin turgor, mucous membrane moisture, and neurological examination to detect any signs of hypernatremia or dehydration.
Initial laboratory tests are crucial to confirm polyuria and assess serum and urine osmolality and electrolytes. These baseline measurements provide essential clues for differential diagnosis.
Water Deprivation Test
The water deprivation test is a cornerstone in the diagnosis of diabetes insipidus. It assesses the body’s ability to concentrate urine in response to fluid restriction and exogenous ADH administration. The traditional indirect water deprivation test involves:
- Baseline Measurements: Obtain baseline urine osmolality, plasma sodium, and plasma osmolality.
- Fluid Deprivation: Instruct the patient to abstain from fluids for a specified period, typically up to 17 hours, or until certain endpoints are reached (plasma sodium ≥ 150 mmol/L or 3-5% body weight loss).
- Hourly Urine Collection: Measure urine volume and osmolality hourly during the deprivation period.
- Desmopressin (DDAVP) Administration: Administer synthetic ADH (desmopressin, DDAVP), usually intranasally or intravenously.
- Post-DDAVP Urine Osmolality: Measure urine osmolality again 1-2 hours after DDAVP administration.
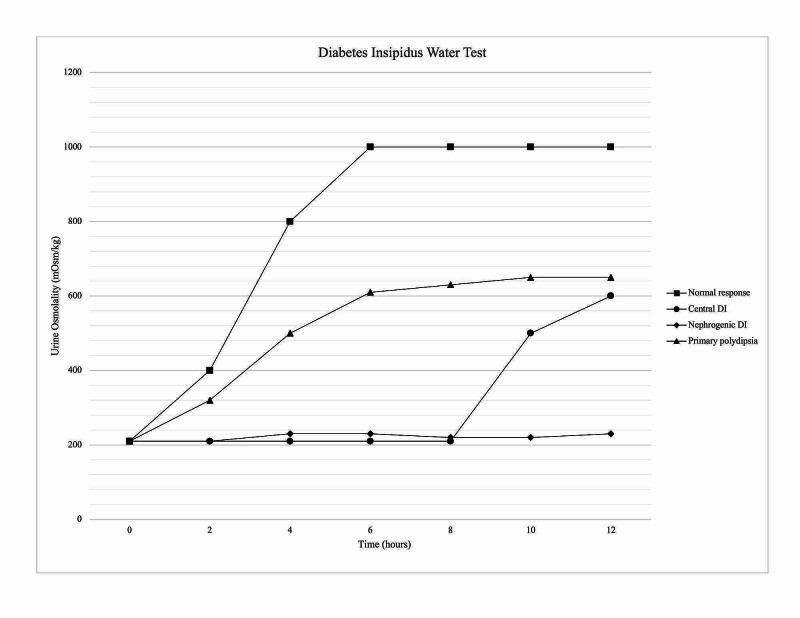
Interpreting Water Deprivation Test Results:
- Normal Response: In healthy individuals, urine osmolality will rise above 800 mOsm/kg during water deprivation, and there will be minimal or no further increase after DDAVP administration.
- Central DI: Urine osmolality remains low (typically < 300 mOsm/kg) during water deprivation. After DDAVP, urine osmolality increases by > 50%, indicating responsiveness to ADH and confirming central DI.
- Nephrogenic DI: Urine osmolality remains low during water deprivation, and there is minimal (< 50%) or no significant increase in urine osmolality after DDAVP administration, indicating renal resistance to ADH.
- Dipsogenic DI: Urine osmolality may increase somewhat during water deprivation but usually remains below normal levels. The response to DDAVP is variable and less pronounced than in CDI. Differentiating DDI from mild CDI can be challenging with the traditional water deprivation test alone.
Limitations of the Traditional Water Deprivation Test:
The indirect water deprivation test has limitations, including a diagnostic accuracy of around 70%. It can be particularly challenging to differentiate dipsogenic DI from partial central DI. In pregnant women, prolonged water restriction poses risks to both mother and fetus, making the traditional test less suitable for gestational DI diagnosis.
Copeptin Measurement: A Modern Diagnostic Marker
Copeptin has emerged as a valuable and more accurate diagnostic marker for diabetes insipidus. Copeptin is a peptide co-secreted with ADH from the same precursor protein, pre-provasopressin. It is more stable and easier to measure than ADH itself, making it clinically practical.
Copeptin-Aided Diagnostic Protocol:
- Baseline Copeptin Measurement: Measure baseline plasma copeptin levels.
- High Baseline Copeptin (> 21.4 pmol/L): Suggestive of nephrogenic DI.
- Low Baseline Copeptin (< 21.4 pmol/L): Requires further osmotic stimulation to differentiate between central and dipsogenic DI.
- Osmotic Stimulation with Hypertonic Saline Infusion: If baseline copeptin is low, perform osmotic stimulation using hypertonic saline (3% saline infusion) to raise serum sodium to at least 150 mmol/L.
- Stimulated Copeptin Measurement: Measure copeptin levels after osmotic stimulation.
- Low Stimulated Copeptin (< 4.9 pmol/L): Confirms central DI.
- Normal or Elevated Stimulated Copeptin (> 4.9 pmol/L): Suggests dipsogenic DI.
Advantages of Copeptin Measurement:
- Improved Diagnostic Accuracy: Copeptin-based diagnosis, particularly with hypertonic saline stimulation, demonstrates high accuracy (up to 97%) in differentiating between CDI, NDI, and DDI.
- Faster and Easier Measurement: Copeptin assays are rapid and require only small plasma samples.
- More Direct Assessment of ADH System: Copeptin reflects endogenous ADH production and release more directly than the indirect water deprivation test.
Copeptin in Gestational DI Diagnosis:
While specific copeptin reference ranges for gestational DI are still under investigation, elevated copeptin levels in the third trimester may be associated with increased risk of pregnancy complications like preeclampsia. Copeptin measurement is becoming increasingly relevant in the diagnostic workup of gestational DI.
Direct Arginine Vasopressin (AVP) Measurement
Direct measurement of arginine vasopressin (AVP), or ADH, has been proposed as a more direct diagnostic approach. However, AVP measurement has technical challenges due to its instability and assay difficulties, limiting its clinical utility. Direct AVP measurements are not routinely used in clinical practice for DI diagnosis.
MRI of the Pituitary Gland
Magnetic resonance imaging (MRI) of the pituitary gland can be a valuable adjunct in diagnosing central diabetes insipidus, especially in cases of suspected structural lesions. MRI findings suggestive of CDI include:
- Infundibular Stalk Thickening: Thickening of the pituitary stalk may indicate inflammatory or infiltrative processes.
- Absence of Posterior Pituitary Bright Spot (PPBS): The normal posterior pituitary gland appears as a bright spot on T1-weighted MRI. Absence of PPBS can suggest CDI, although it may be present in early stages.
MRI is particularly useful in identifying underlying causes of CDI, such as tumors, infiltrative diseases, or structural abnormalities in the hypothalamus-pituitary region.
Differential Diagnosis
It’s essential to differentiate diabetes insipidus from other conditions that can cause polyuria and polydipsia, including:
- Diabetes Mellitus: Rule out hyperglycemia and glucosuria as causes of polyuria.
- Primary Polydipsia (Psychogenic Polydipsia): Differentiate dipsogenic DI from psychogenic polydipsia, where excessive water intake is driven by psychological factors rather than a hypothalamic thirst disorder.
- Hypercalcemia and Hypokalemia: These electrolyte imbalances can cause nephrogenic DI-like symptoms.
- Sickle Cell Anemia and Histiocytosis: Rarely, these conditions can affect renal function and cause polyuria.
Table 1. Summary of Diabetes Insipidus Types, Diagnosis, and Management
| Feature | Central DI | Nephrogenic DI | Dipsogenic DI | Gestational DI |
|---|---|---|---|---|
| Cause | ADH deficiency | Renal resistance to ADH | Dysregulated thirst mechanism | Placental vasopressinase-induced ADH degradation |
| Vasopressin Response | Urine osmolality ↑ with DDAVP | Minimal urine osmolality ↑ with DDAVP | Variable response to DDAVP | Urine osmolality ↑ with DDAVP |
| Diagnosis | Water deprivation test + DDAVP, Copeptin, MRI | Water deprivation test + DDAVP, Copeptin | Water deprivation test, Copeptin, Clinical History | Serum/Urine Osmolality, Clinical History |
| Management | DDAVP, Thiazide diuretics, Fluid management | Treat underlying cause, Thiazide diuretics, Renal diet, Fluid management | Behavioral therapy, Antipsychotics (if needed), Fluid management | DDAVP, Fluid management |
Alt text: Summary table of Diabetes Insipidus types: comparing Central DI, Nephrogenic DI, Dipsogenic DI, and Gestational DI on Description, Causes, Vasopressin Response, Diagnosis, and Clinical Management.
Figure 7. Urine Osmolality Response in Water Deprivation Test for DI Types
Alt text: Graph illustrating urine osmolality during water deprivation test: comparing normal response, Central DI response with significant increase after DDAVP, and Nephrogenic DI showing minimal response to DDAVP.
Conclusion: Advancing Diabetes Insipidus Diagnosis
Accurate and timely diagnosis of diabetes insipidus is essential for effective management and improving the quality of life for affected individuals. An up-to-date diagnostic approach integrates clinical evaluation, refined water deprivation testing, and importantly, copeptin measurements. Copeptin has emerged as a robust biomarker, enhancing diagnostic accuracy, particularly in differentiating between the various types of DI. While the water deprivation test remains a valuable tool, copeptin-aided protocols offer a more precise and clinically practical diagnostic pathway. For gestational DI, clinical history and serum/urine osmolality are key, with copeptin research evolving. Continued advancements in diagnostic strategies, including the integration of copeptin and pituitary MRI, are refining our ability to diagnose diabetes insipidus with greater accuracy and guide tailored management strategies for each subtype of this challenging disorder. Early and precise diagnosis, coupled with appropriate treatment, can significantly mitigate the impact of diabetes insipidus and improve patient outcomes.
References
[1] Christ-Crain M, Winzeler B, Refardt J. Diabetes insipidus. In: Feingold KR, Anawalt B, Boyce A, et al., editors. Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc.; 2000-. 2018 Jul 21. Available from: https://www.ncbi.nlm.nih.gov/books/NBK279081/
[2] Fenske WK, Allolio B. Clinical review: Current state and future perspectives in the diagnosis of diabetes insipidus: A clinical review. J Clin Endocrinol Metab. 2012 Oct;97(10):3425-37. doi: 10.1210/jc.2012-1008. Epub 2012 Jul 25. PMID: 22833637.
[3] Verbalis JG, Goldsmith SR, Greenberg A, et al. Hyponatremia treatment guidelines 2013: expert panel recommendations from the American College of Endocrinology and the Endocrine Society. J Clin Endocrinol Metab. 2013 Apr;98(4):1141-3. doi: 10.1210/jc.2013-1099. PMID: 23576765.
[4] Robertson GL. Regulation of vasopressin secretion. Front Neuroendocrinol. 1995 Apr;16(2):131-53. doi: 10.1006/frne.1995.1005. PMID: 7627991.
[5] Weissberger AJ, Major PW, Pringle MA, Bowden CJ, Lavercombe PS, Buchanan CR. Traumatic diabetes insipidus: early and late onset. Aust N Z J Med. 1992 Feb;22(1):45-51. doi: 10.1111/j.1445-5994.1992.tb00408.x. PMID: 1575533.
[6] Repaske DR. Vasopressin gene mutations and inherited diabetes insipidus. J Clin Endocrinol Metab. 1994 Apr;78(4):728-33. doi: 10.1210/jcem.78.4.7908284. PMID: 7908284.
[7] Fonseca SG, Fukuma N, Lipson KL, et al. WFS1 gene mutations in Wolfram syndrome downregulate secretion of insulin, IAPP, and GLP-1. J Biol Chem. 2005 Aug 26;280(30):27269-77. doi: 10.1074/jbc.M504343200. Epub 2005 Jun 15. PMID: 15955781.
[8] Osman E, Ma J, Rosendahl AH, et al. Wolframin modulates endoplasmic reticulum stress and calcium homeostasis in INS-1E insulinoma cells. Endocrinology. 2003 Nov;144(11):5102-11. doi: 10.1210/en.2003-0282. PMID: 12960043.
[9] Barrett TG, Bundey SE. Wolfram (DIDMOAD) syndrome. J Med Genet. 1997 Dec;34(12):838-41. doi: 10.1136/jmg.34.12.838. PMID: 9489945; PMCID: PMC1051144.
[10] Muhsin SA, Mount DB. Drug-induced electrolyte abnormalities. Nat Clin Pract Nephrol. 2006 Nov;2(11):637-47. doi: 10.1038/ncpneph0323. PMID: 17106444.
[11] Christ-Crain M, Bichet DG, Fenske WK, et al. Diabetes insipidus. Nat Rev Dis Primers. 2019 Aug 22;5(1):54. doi: 10.1038/s41572-019-0103-2. PMID: 31431616.
[12] Gradus-Pizlo I, Kostrzewa G, Gałecki P, Talarowska M. Clozapine-induced nephrogenic diabetes insipidus – a case report and literature review. Psychiatr Pol. 2017 Dec 30;51(6):1147-1157. doi: 10.12740/PP/OnlineFirst/64488. Epub 2017 Dec 29. PMID: 29293814.
[13] Verbalis JG. Disorders of body water homeostasis. Best Pract Res Clin Endocrinol Metab. 2003 Dec;17(4):471-503. doi: 10.1016/S1521-690X(03)00055-1. PMID: 14654238.
[14] Sailer CO, Winzeler B, Christ-Crain M. Primary polydipsia. J Clin Endocrinol Metab. 2017 Aug 1;102(8):3141-3148. doi: 10.1210/jc.2017-00529. PMID: 28838097.
[15] Durr JA, Hoggard JG, Hunt JM, Alonso-Vazquez P, Patak EM, Zerbe RL. Vasopressinase of pregnancy: degradation of vasopressin and a[9-desglycinamide]vasopressin, and development of a radioimmunoassay. Endocrinology. 1992 Jun;130(6):3771-8. doi: 10.1210/endo.130.6.1600751. PMID: 1600751.
[16] Christ-Crain M, Fenske WK. Copeptin in diabetes insipidus. Endocrinol Metab Clin North Am. 2017 Dec;46(4):943-955. doi: 10.1016/j.ecl.2017.07.009. PMID: 29080605.
[17] Timper K, Fenske W, Kühn F, et al. Diagnostic accuracy of copeptin in the differential diagnosis of the polyuria-polydipsia syndrome: a prospective multicenter study. J Clin Endocrinol Metab. 2015 May;100(5):2268-74. doi: 10.1210/jc.2014-4356. Epub 2015 Mar 6. PMID: 25742431.
[18] Adrogué HJ, Madias NE. Hyponatremia. N Engl J Med. 2000 May 25;342(21):1581-9. doi: 10.1056/NEJM200005253422107. PMID: 10824077.
[19] Sterns RH, Riggs JE, Schochet SS. Osmotic demyelination syndrome following rapid correction of hyponatremia. N Engl J Med. 1986 Nov 27;315(24):1535-42. doi: 10.1056/NEJM198611273152406. PMID: 3785399.
[20] Mount DB. Clinical disorders of water homeostasis. N Engl J Med. 2007 Aug 23;357(6):575-84. doi: 10.1056/NEJMra070527. PMID: 17699030.
[21] Ganong WF. Review of medical physiology. 11th ed. Norwalk, Conn: Appleton & Lange; 1993.
[22] Scherbaum WA, Bottazzo GF, Pozzilli P, Doniach D. Autoantibodies to vasopressin cells in idiopathic diabetes insipidus. Lancet. 1984 Nov 10;2(8412):1173-6. doi: 10.1016/s0140-6736(84)90672-5. PMID: 6149573.
[23] Miller M, Moses AM. Clinical states due to alteration in ADH release and action. In: DeGroot LJ, Jameson JL, eds. Endocrinology. 4th ed. Philadelphia: WB Saunders; 2001.
[24] Fenske WK, Quinkler M, Lorenz D, et al. Copeptin in the differential diagnosis of hyponatremia. J Clin Endocrinol Metab. 2011 Jul;96(7):1915-25. doi: 10.1210/jc.2011-0423. Epub 2011 May 18. PMID: 21593156.
[25] Zerbe RL, Robertson GL. A comparison of plasma vasopressin measurements with a standard indirect test in the differential diagnosis of polyuria. N Engl J Med. 1981 Jun 18;305(25):1539-46. doi: 10.1056/NEJM198112173052503. PMID: 7300821.
[26] Robertson GL, Aycinena P, Zerbe RL. Osmotic and nonosmotic control of vasopressin function. Recent Prog Horm Res. 1976;32:225-72. PMID: 185834.
[27] Morgenthaler NG, Struck J, Alonso C, Bergmann A. Assay for the measurement of copeptin, a stable peptide derived from the precursor of vasopressin. Clin Chem Lab Med. 2006;44(1):51-7. doi: 10.1515/CCLM.2006.010. PMID: 16393147.
[28] Christ-Crain M, Oppermann C, Fenske W, et al. Copeptin, a biomarker of absolute or relative arginine vasopressin deficiency, in polyuria-polydipsia syndrome. J Clin Endocrinol Metab. 2010 Dec;95(12):5298-305. doi: 10.1210/jc.2010-1445. Epub 2010 Oct 6. PMID: 20926527.
[29] Bichet DG. Clinical and laboratory diagnosis of nephrogenic diabetes insipidus. Kidney Int Suppl. 1994 Jun;45:S77-81. PMID: 8067482.
[30] Miller M, Dalakos T, Moses AM, Fellerman H, Streeten DH. Recognition of partial defects in antidiuretic hormone secretion. Ann Intern Med. 1970 Apr;72(5):721-9. doi: 10.7326/0003-4819-72-5-721. PMID: 5468571.
[31] Ishikawa SE, Saito T, Yoshida S. The effect of hydrochlorothiazide and indomethacin on urine osmolality and plasma vasopressin concentration in patients with partial central diabetes insipidus. J Clin Endocrinol Metab. 1985 Jul;61(1):22-9. doi: 10.1210/jcem-61-1-22. PMID: 4004399.
[32] Kortenoeven ML, Cutler C, Jørgensen CB, Fenton RA. Thiazides in nephrogenic diabetes insipidus: mechanism of action. Am J Physiol Renal Physiol. 2015 Feb 15;308(4):F434-45. doi: 10.1152/ajprenal.00529.2014. Epub 2014 Dec 17. PMID: 25519668; PMCID: PMC4344637.
[33] Procino G, Sangenario R, Gesualdo L, Schena FP, Sallustio F. Statins and nephrotic syndrome: atorvastatin increases aquaporin-2 expression in puromycin-induced nephropathy. J Nephrol. 2012 Mar-Apr;25(2):231-8. doi: 10.5301/jn.5000004. Epub 2011 Jun 23. PMID: 21706375.
[34] Garzotto F, Verlato G, D’Alo’ F, et al. Atorvastatin for lithium-induced nephrogenic diabetes insipidus: a double-blind, randomized, placebo-controlled, pilot trial. J Nephrol. 2021 Mar;34(2):407-415. doi: 10.1007/s40620-020-00826-x. Epub 2020 Nov 28. PMID: 33245564; PMCID: PMC7933254.
[35] Davison JM, Sheills EA, Philips N, Barron WM. Metabolic clearance of desmopressin during and after pregnancy. Am J Obstet Gynecol. 1997 Nov;177(5):1228-33. doi: 10.1016/s0002-9378(97)70234-8. PMID: 9398117.
[36] Wagner G, Bohr VA, Nørgaard JP, Pedersen EB. DDAVP in pregnant women with central diabetes insipidus. Acta Obstet Gynecol Scand. 1995 Mar;74(3):175-9. doi: 10.3109/00016349509024455. PMID: 7717228.
[37] Kuno K, Okada M, Morita H, et al. Oligohydramnios in a pregnant woman with gestational diabetes insipidus treated with desmopressin. Gynecol Obstet Invest. 2004;57(1):53-5. doi: 10.1159/000075127. PMID: 14634281.
[38] Jørgensen JS, Feldt-Rasmussen B, Wagner G, Rittig S, Nørgaard JP. Gestational diabetes insipidus and type 2 diabetes mellitus: are they related? Obstet Med. 2012 Jun;5(2):68-70. doi: 10.1258/om.2012.120024. PMID: 27516837; PMCID: PMC4995310.
[39] Verhaeghe J, Van Bree R, Van Herck E, et al. Sexual dimorphism in placental vasopressinase activity: higher activity in placentas of male fetuses. J Clin Endocrinol Metab. 1995 Jun;80(6):2754-7. doi: 10.1210/jcem.80.9.7629246. PMID: 7629246.