Introduction
Duchenne muscular dystrophy (DMD) is a devastating X-linked recessive genetic disorder characterized by progressive muscle weakness and degeneration. This condition arises from mutations in the dystrophin gene, leading to the absence or insufficiency of functional dystrophin, a crucial protein for muscle fiber integrity and function. The reported prevalence of DMD varies slightly geographically, with approximately 15.9 cases per 100,000 live male births in the USA and 19.5 cases per 100,000 in the UK. Individuals with DMD experience ongoing muscle damage, resulting in a cascade of debilitating symptoms including motor delays, loss of ambulation, respiratory compromise, and cardiomyopathy. While the progression of skeletal and cardiac muscle involvement can differ among individuals, the prognosis remains severe, with death typically resulting from cardiac or respiratory failure.
This article represents the first part of a three-part series designed to update the 2010 Duchenne muscular dystrophy care considerations. This updated guidance is supported by the US Centers for Disease Control and Prevention (CDC), in collaboration with the TREAT-NMD network, the Muscular Dystrophy Association, and Parent Project Muscular Dystrophy. The impetus for this update stems from significant advancements in DMD care, notably improved patient survival rates due to multidisciplinary approaches, and evolving diagnostic and therapeutic strategies across various medical specialties. This progress has shifted the focus towards anticipatory care, emphasizing the prevention, early detection, and management of predictable disease complications to enhance patient outcomes and quality of life.
Furthermore, the increasing life expectancy of individuals with DMD underscores the critical need to address quality of life issues and psychosocial well-being. A crucial aspect of this updated guidance is the improved coordination of care during the transition from pediatric to adult healthcare systems. Finally, the rapid evolution of DMD therapies, including both established treatments and promising emerging genetic and molecular approaches, necessitates a re-evaluation of care considerations. Specifically, new insights into the effectiveness, side effects, and limitations of glucocorticoids have emerged, alongside the urgent need to identify reliable biomarkers and outcome measures for assessing novel therapies.
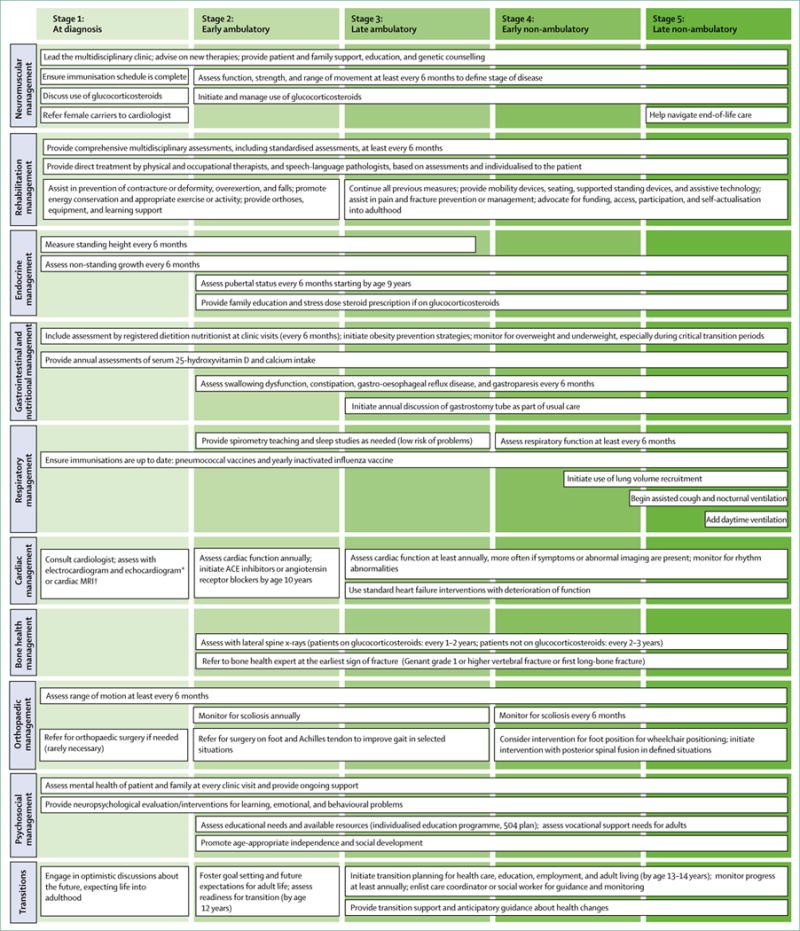
Part 1 of this comprehensive review focuses on key aspects of diagnosis and management, including: the diagnostic process for DMD, neuromuscular management strategies, rehabilitation approaches, endocrine management (addressing growth, puberty, and adrenal insufficiency), and gastrointestinal management (encompassing nutrition and dysphagia). Subsequent parts of this series will delve into other crucial domains such as psychosocial support, primary and emergency care, and the complex process of care transitions across the lifespan. Figure 1 provides a visual overview of the comprehensive, multidisciplinary care framework recommended for individuals with Duchenne muscular dystrophy, organized by disease stage and relevant topic areas.
Figure 1. Comprehensive care of individuals with Duchenne muscular dystrophy across disease stages.
Care for Duchenne muscular dystrophy patients requires a team of specialists led by a neuromuscular expert. This figure outlines assessments and interventions across all disease stages and topics in this review series. *Echocardiogram is recommended for patients aged 6 years and younger. †Cardiac MRI is recommended for patients older than 6 years.
Diagnosis of Duchenne Muscular Dystrophy
A prompt and accurate diagnosis of Duchenne muscular dystrophy is paramount for initiating timely care and improving patient outcomes. While the fundamental diagnostic approach for DMD remains consistent with the 2010 guidelines, ongoing refinements and increased awareness are crucial for minimizing diagnostic delays. The diagnostic journey typically begins in early childhood when parents or caregivers observe suggestive signs and symptoms. These early indicators often include generalized weakness, clumsiness, the presence of a Gowers’ sign (using hands to “walk up” the legs when rising from the floor), difficulty climbing stairs, or toe walking. Prompt referral to a neuromuscular specialist, ideally in collaboration with a geneticist or genetic counselor, is essential to expedite the diagnostic process and prevent unnecessary delays.
In some cases, DMD diagnosis may be considered when a child presents with developmental delays, particularly in motor milestones. Elevated levels of serum enzymes, such as alanine aminotransferase (ALT), aspartate aminotransferase (AST), lactate dehydrogenase (LDH), or creatine kinase (CK), detected during routine bloodwork, can also raise suspicion for DMD. It is important to note that elevated liver enzymes (ALT, AST, LDH) can sometimes misleadingly point towards hepatic dysfunction, potentially delaying the correct diagnosis of DMD. Therefore, in cases of unexplained elevated liver enzymes in young boys, particularly in conjunction with motor concerns, DMD should be considered as part of the differential diagnosis.
Figure 2. Diagnostic algorithm for Duchenne muscular dystrophy, outlining genetic and muscle biopsy testing.
Early signs and symptoms of DMD are based on clinical observations by Ciafaloni and colleagues. DMD=Duchenne muscular dystrophy.
Genetic Testing: Genetic testing plays a central role in confirming a DMD diagnosis. Approximately 70% of individuals with DMD harbor deletions or duplications of one or more exons within the dystrophin gene. Therefore, initial genetic testing typically involves dystrophin gene deletion and duplication analysis. Multiplex ligation-dependent probe amplification (MLPA) or comparative genomic hybridization (CGH) array are the preferred methods for this analysis, as standard multiplex PCR techniques may fail to detect duplications. Identifying the precise boundaries of a deletion or duplication mutation using MLPA or CGH array can provide valuable information regarding the potential impact on the dystrophin reading frame, helping to predict the severity of the mutation.
If initial deletion and duplication testing is negative, further genetic sequencing is necessary to screen for other types of dystrophin gene mutations, which account for approximately 25–30% of DMD cases. These include point mutations (nonsense or missense mutations), small deletions, and small duplications or insertions. Next-generation sequencing (NGS) technologies are well-suited for identifying these less common mutation types.
Muscle Biopsy: In cases where clinical suspicion for DMD remains high despite negative genetic testing, a muscle biopsy is indicated. Muscle biopsy allows for direct assessment of dystrophin protein levels within muscle tissue. Immunohistochemistry on muscle cryosections or Western blot analysis of muscle protein extracts are used to detect the presence and quantity of dystrophin protein. The absence or significant reduction of dystrophin confirms the diagnosis of DMD in these genetically unresolved cases.
Female Carriers: Genetic counseling is crucial for families of individuals diagnosed with DMD to identify female relatives at risk of being carriers of the dystrophin gene mutation. Carrier testing is strongly recommended for female relatives of a male with genetically confirmed DMD. For child relatives, ethical guidelines from organizations like the American Medical Association regarding genetic testing in children should be carefully considered and followed. Identified female carriers have important reproductive options to consider, including preimplantation genetic diagnosis (PGD) or prenatal genetic testing via chorionic villus sampling (CVS) or amniocentesis. Furthermore, female carriers are not simply asymptomatic gene carriers; they require ongoing medical assessment and follow-up, particularly for cardiac health, as detailed in part 2 of this review series which focuses on cardiac management.
Newborn Screening: The concept of newborn screening for DMD has been explored since the mid-1970s, primarily through creatine kinase (CK) measurement from dried blood spots. A two-tiered newborn screening system has been proposed and tested, where samples with elevated CK levels undergo subsequent dystrophin gene mutation analysis. While newborn screening studies for DMD have been conducted in several countries, many have been discontinued, and DMD is not currently part of the Recommended Uniform Screening Panel in the United States. This panel largely focuses on neonatal-onset disorders where early treatment demonstrably improves outcomes. However, renewed interest in newborn screening for DMD is growing, driven by stakeholder support and the understanding that emerging DMD therapies may be most effective when initiated presymptomatically, before irreversible muscle damage occurs. Early diagnosis through newborn screening could pave the way for proactive therapeutic interventions and potentially alter the disease trajectory in DMD.
Neuromuscular Management of Duchenne Muscular Dystrophy
Following a confirmed diagnosis of DMD, a neuromuscular specialist assumes the role of lead clinician, orchestrating and overseeing the comprehensive care plan for the individual throughout their lifespan. The neuromuscular specialist is uniquely positioned to guide patients and their families through the increasingly complex landscape of DMD diagnostics and therapeutics. Their responsibilities encompass a wide spectrum of roles, as detailed in Panel 1.
Assessments: Regular and standardized clinical assessments of neuromuscular function are the bedrock of effective DMD management. The assessment tools outlined in the 2010 care considerations remain relevant and valuable. Clinics should utilize a core set of tests with which they are familiar and whose clinical significance they understand. Crucially, a multidisciplinary team approach is essential to ensure consistency in assessments and avoid redundant testing. Suggested assessments are detailed in the Appendix and are further discussed in the Rehabilitation Management section. Recent research has highlighted the importance of minimum clinically important differences (MCID), the predictive capacity of standardized functional assessments, and the ranges of optimal responsiveness for these tests. These findings underscore the ongoing value of standardized functional assessments in monitoring disease progression and evaluating treatment efficacy across the patient’s life. Moreover, newer assessment tools are being developed and validated to guide the management of older, non-ambulatory individuals, emphasizing the need for continuous clinical testing throughout the lifespan in DMD.
Interventions: Physiotherapy, as discussed in the Rehabilitation Management section, and glucocorticoid therapy remain the cornerstone interventions in DMD management. These treatments should be initiated promptly after diagnosis and continued even after loss of ambulation to maximize their benefits. Figure 3 outlines key considerations for glucocorticoid initiation and use in DMD. Long-term glucocorticoid therapy has been consistently shown to delay the loss of ambulation, preserve upper limb and respiratory function, and reduce the need for scoliosis surgery. Recent studies further support the benefits of initiating glucocorticoids in younger children, before significant muscle decline has occurred. An ongoing clinical trial is investigating the efficacy of weekend glucocorticoid dosing in boys younger than 30 months, which may provide further insights into optimal dosing strategies.
Despite the well-established benefits of glucocorticoid therapy, uncertainties persist regarding the optimal type of glucocorticoid and dosage. This lack of clarity can increase the risk of both undertreatment and overtreatment, potentially confounding the interpretation of clinical trials evaluating novel therapies. Large-scale natural history and cohort studies have demonstrated that glucocorticoid treatment prolongs ambulation. The mean age of ambulation loss is approximately 10.0 years in individuals receiving less than one year of corticosteroid treatment, 11.2 years in those treated with daily prednisone, and 13.9 years in those taking daily deflazacort. Some studies suggest that weekend-only prednisone dosing may achieve similar efficacy to daily dosing. A phase 3 double-blind randomized controlled trial (RCT) comparing deflazacort at 0.9 mg/kg per day, deflazacort at 1.2 mg/kg per day, prednisone at 0.75 mg/kg per day, and placebo demonstrated that all treatment groups showed improved muscle strength compared to placebo. Deflazacort was associated with less weight gain than prednisone in this study. Ongoing research continues to investigate the benefit-risk profile of deflazacort compared to prednisone in DMD.
Figure 3. Care considerations for initiating and managing glucocorticoid (steroid) therapy in Duchenne muscular dystrophy.
ACTH=adrenocorticotropic hormone. CRH=corticotropin-releasing hormone. HPA=hypothalamic-pituitary-adrenal.
Emerging Treatments: The landscape of DMD drug development has dramatically shifted since the 2010 care considerations. The pipeline of potential DMD therapies is constantly evolving, and comprehensive, updated information on clinical trials is readily available at ClinicalTrials.gov and the WHO International Clinical Trials Registry Platform. DMD, as a rare disease, presents unique challenges for clinical trial capacity. The increasing number of DMD trials strains the ability to recruit sufficient patients who meet specific eligibility criteria. This underscores the urgent need to optimize patient recruitment strategies and promote trial readiness initiatives, such as patient registries, the identification of clinically meaningful outcome measures, and robust natural history studies.
In August 2014, ataluren received conditional marketing authorization from the European Commission for use in the European Union. Ataluren targets approximately 11% of boys with DMD whose condition is caused by a nonsense mutation (stop codon) in the dystrophin gene. In September 2016, the US Food and Drug Administration (FDA) granted accelerated approval for eteplirsen. Eteplirsen targets approximately 13% of boys with a dystrophin gene mutation amenable to exon 51 skipping. Ataluren and eteplirsen represent the first in a wave of mutation-specific therapies to achieve regulatory approval for DMD. Additional dystrophin restoration therapies are in various stages of development, with some approaching or undergoing regulatory review. The FDA has also granted full approval for deflazacort, marking it as the first glucocorticoid specifically labeled for DMD treatment.
Beyond dystrophin-targeted therapies, other drug classes are under investigation in clinical trials for DMD. These include drugs targeting myostatin (a negative regulator of muscle growth), anti-inflammatory and antioxidant molecules, compounds designed to reduce fibrosis, drugs to enhance vasodilatation, agents to improve mitochondrial function, and drugs to upregulate utrophin (a dystrophin-related protein). However, it is crucial to emphasize that without completed clinical studies and regulatory approval, none of these investigational drugs can be prescribed for individuals with DMD outside of a clinical trial setting.
Rehabilitation Management in Duchenne Muscular Dystrophy
Duchenne muscular dystrophy is characterized by a predictable pattern of progressive muscle degeneration and weakness. This leads to postural adaptations, an increased risk of contractures and deformities, and functional limitations stemming directly from dystrophin deficiency. Advances in DMD management have resulted in prolonged ambulation, reduced incidence of severe contractures and scoliosis, and extended functional capacity and participation in various life domains. The rehabilitation team for individuals with DMD is multidisciplinary, encompassing physicians, physical therapists, occupational therapists, speech-language pathologists, orthotists, and durable medical equipment providers. Panel 2 and the Appendix provide a comprehensive overview of recommended rehabilitation assessments and interventions. Effective rehabilitation management requires a deep understanding of DMD pathology, pathokinesiology, natural history, and disease progression. Rehabilitation plans should be individualized, considering each person’s goals and lifestyle to maximize their quality of life throughout their lifespan. From the time of diagnosis onwards, assessment and anticipatory management should address all domains of the International Classification of Functioning, Disability and Health (ICF) framework. The overarching goals are to minimize contractures, deformities, functional loss, skin integrity issues, pain, and cardiorespiratory compromise.
Assessments: Multidisciplinary rehabilitation assessments are crucial for monitoring disease progression and guiding interventions. These assessments include evaluating passive range of motion, muscle extensibility, posture and alignment, strength, functional abilities, quality of life, and participation in daily activities. Panel 2 and the Appendix detail specific assessments. Specialized functional assessments involve analyzing movement patterns and utilizing standardized tools specific to DMD and other neuromuscular disorders. The North Star Ambulatory Assessment (NSAA) and timed function tests are fundamental clinical assessments during the ambulatory phase and should be performed every 6 months. These assessments demonstrate high validity and reliability, strong correlations over time, established minimum clinically important differences, and predictive capabilities for functional motor changes. This makes them invaluable for tracking disease progression and evaluating the effectiveness of new therapies. Identifying optimally responsive test ranges further enhances their predictive accuracy. For example, in the 6-minute walk test (6MWT), understanding the interplay between age, baseline distance, and genetics can refine research design and inform clinical care.
Predicting functional changes in clinical practice must consider the individual’s current capabilities, limitations of effort-based assessments, potential confounding factors like contractures, and genetic background. Tests that can anticipate upcoming functional changes are essential for proactive care planning, including timely implementation of impairment-level interventions and anticipating future equipment needs. Specifically, before the age of 7 years, gains may be observed in the 6MWT and timed function tests. After age 7, certain performance thresholds in these tests are associated with a higher risk of functional decline in ambulation within the following 12 months. These thresholds include a 6MWT distance less than 325 meters, time to stand greater than 30 seconds, time to climb four stairs greater than 8 seconds, 10-meter walk or run time exceeding 10–12 seconds, and a mean linearized NSAA score of 34 or less (raw score of nine). Functional assessment also encompasses evaluating activities of daily living (ADLs) and determining the need for adaptive equipment or assistive technology. Various validated tools can be employed to assess quality of life in individuals with DMD.
The increasing potential for early DMD diagnosis through newborn screening and the emergence of early-intervention therapies highlight the timeliness of standardized testing in infants and young children with DMD. The Bayley Scales of Infant Development-III and Griffiths Mental Development Scales are valuable tools for measuring developmental progress in young children and can identify early developmental delays in DMD. The NSAA, with modifications, can be used in children as young as 3 years old. Hip kinematics during gait are clinically meaningful outcome measures in children aged 4–8 years. Other measures assessing antigravity function, considered exploratory in DMD, include the Alberta Infant Motor Scale, Hammersmith Functional Motor Scale Expanded, and the Gross Motor Function Measure. Early assessment and intervention for learning, attention, and sensory processing differences are also crucial, beginning in young children. In older, non-ambulatory individuals, the Brooke Upper Extremity Scale, Egen Klassifikation scale, and elbow flexion and grip strength are responsive to changes over 1–2 years. Assessments in this population now also include reachable workspace and upper limb function tests like the Performance of Upper Limb (PUL) test.
Consistent use of the same functional measures within individual clinics is recommended to effectively track changes over time. Newer assessments should be incorporated as appropriate. Rehabilitation specialist assessments are recommended at least every 4–6 months throughout life, with more frequent assessments triggered by clinical concerns, changes in functional status, or specific needs.
Interventions: Direct physical, occupational, and speech and language therapy should be provided in outpatient, school, and home settings, and should continue throughout adulthood. These therapies are often augmented by interventions provided during hospital admissions. Panel 2 and the Appendix provide a detailed overview of rehabilitation interventions.
Prevention of Contracture and Deformity: Managing muscle extensibility and joint mobility is crucial for preventing or minimizing contractures and deformities. Contractures develop due to the inability to move joints through their full range of motion, prolonged static positioning, muscle imbalances around joints, and fibrotic changes in muscles. Restricted breathing patterns and fibrosis of intercostal muscles can also limit chest wall mobility. Maintaining passive range of motion, muscle extensibility, chest wall mobility, and symmetry is essential for optimizing movement, functional positioning, maintaining ambulation, preventing fixed contractures and deformities, optimizing respiratory function, and preserving skin integrity. Musculoskeletal management requires a collaborative team approach involving neuromuscular specialists, physical therapists, occupational therapists, rehabilitation physicians, orthotists, and orthopedic surgeons.
Preventing contractures and deformities necessitates daily passive stretching of joints, muscles, and soft tissues at risk of tightness. Interventions also include supporting movement by reducing the effects of gravity and optimizing biomechanics to facilitate active movement. Manual therapy techniques and prolonged soft tissue elongation, along with optimal positioning strategies, including individualized splinting, orthotic interventions, standing devices, serial casting, and customized seating and power positioning components in mobility devices, are also crucial. A daily preventive home stretching program, initiated under the guidance of physical and occupational therapists, should begin before passive range of motion is lost. Stretching should target areas known to be prone to contractures or deformities (Panel 2). Regular stretching of the ankles, knees, and hips should commence soon after diagnosis and continue into adulthood. Upper extremity stretching becomes particularly important after loss of ambulation.
The Appendix provides a comprehensive overview of care considerations related to various assistive and mobility devices, including ankle-foot orthoses (AFOs), knee-ankle-foot orthoses (KAFOs), serial casting, standing devices, and manual and motorized mobility devices. Power stand-and-drive motorized wheelchairs are increasingly used as alternatives to KAFOs for standing mobility. While KAFOs may still be appropriate in certain situations, they are often viewed as therapeutic tools to supplement, rather than replace, motorized mobility. Technological innovations, ranging from simple adaptive aids (e.g., elevated lap trays, adaptive straws) to advanced technologies (e.g., robotics, Bluetooth-enabled devices, infrared environmental controls, smartphones, tablets, computers, voice activation systems), can significantly enhance function and independence. Adaptive equipment and home modifications may include patient lifts for safe transfers, ramps, stair lifts, bathroom modifications, specialized beds and mattresses, and vehicle adaptations. Personal care attendants can also play a vital role in maximizing independence and participation.
Exercise and Physical Activity: Physical therapists prescribe, monitor, and guide exercise programs to prevent sedentary lifestyles and associated problems like social isolation and overweight. However, the impact of exercise on muscle degeneration in dystrophinopathies is complex and not fully understood. Potential risks include muscle damage due to structural fragility, metabolic abnormalities, nitric oxide imbalances contributing to ischemia during exercise, and reduced exercise capacity. Eccentric muscle activity and high-resistance strength training should be avoided. Submaximal aerobic exercise or activity, with careful attention to avoiding overexertion and ensuring adequate rest, is generally recommended, particularly in the early stages of the disease. Swimming is highly recommended from the early ambulatory phase and can often be continued into adulthood. Cycling, particularly assisted cycling and robotic-assisted movement, is also recommended as a submaximal aerobic activity and can be continued into adulthood. Appropriate adaptive equipment and assistive technology can facilitate safe physical activity.
Pain Management: Pain assessment and management are essential for individuals with DMD at all ages. Comprehensive team management is required, encompassing physical therapy, postural correction, orthotic interventions, splinting, wheelchair and bed modifications to enable independent weight shifting and pressure relief, and pharmacological approaches. Back pain, especially in the context of glucocorticoid treatment, should prompt evaluation for vertebral fractures. Fracture prevention and management are discussed in detail in part 2 of this review series.
Endocrine Management in Duchenne Muscular Dystrophy
Endocrine complications are common in DMD and its treatment, including impaired growth, delayed puberty, and adrenal insufficiency. The goals of endocrine management are to monitor growth and development, identify and diagnose hormone deficiencies, provide hormone replacement therapy when indicated, and prevent life-threatening adrenal crises. While expert opinion papers and reviews exist, robust data on the safety and efficacy of growth hormone and testosterone therapy in DMD are limited. The following care considerations are based on evidence and experience from using these therapies in other conditions, adapted for DMD. Figure 4 provides an overview of endocrine assessments and interventions.
Figure 4. Endocrine assessments and interventions for growth and puberty in Duchenne muscular dystrophy.
Growth: Impaired linear growth is frequent in DMD and can be exacerbated by glucocorticoid treatment. Linear growth should be monitored every 6 months until puberty completion and attainment of final adult height. Standing height is the preferred measure for ambulatory individuals. Height should be plotted on standardized growth curves. For non-ambulatory individuals, regular assessment using non-standing height measures should begin during the ambulatory phase to ensure accurate growth monitoring after ambulation loss. Arm span, ulnar length, tibia length, knee height, and segmentally measured recumbent length have all been used, but none are validated specifically for DMD, and all require specialized training or equipment. Each institution should select and consistently use the measure that best fits their clinical setting.
A decline in growth trajectory, indicated by crossing down through height percentiles or an annualized height velocity less than 4 cm per year, suggests impaired linear growth and warrants referral to an endocrinologist. Referral is also indicated for individuals with height below the third percentile, regardless of growth trajectory. Assessment of impaired linear growth should include standard screening tests for endocrine hormone or other abnormalities associated with growth failure. Data on the safety and efficacy of recombinant human growth hormone in DMD are limited. One retrospective study showed short-term benefit on height velocity, but some boys experienced side effects like intracranial hypertension, glucose intolerance, and scoliosis progression. Published studies have not followed patients to final height, and no large studies have definitively established whether growth hormone therapy negatively impacts muscle function or causes other adverse effects. Theoretical concerns exist that increased height could be detrimental to muscle function in DMD. Currently, routine use of recombinant human growth hormone for DMD-related growth failure is not recommended. The decision to treat should be based on a thorough discussion of potential risks and benefits and preferably reserved for individuals with abnormal growth hormone stimulation test results.
Puberty: Delayed puberty due to hypogonadism is a potential complication of glucocorticoid therapy and can be psychologically distressing, negatively impacting quality of life. Absence of pubertal development by age 14 years necessitates prompt referral to an endocrinologist. Biochemical testing using appropriate pediatric or ultrasensitive assays should confirm hypogonadism in individuals with delayed puberty. A left-hand radiograph to assess bone age should also be considered.
Testosterone replacement therapy is recommended for confirmed hypogonadism in patients older than 14 years and can be considered in boys older than 12 years on glucocorticoids with absent pubertal development. While clinical trials specifically assessing testosterone use in DMD are lacking, it is standard care for pathological pubertal delay in pediatrics and glucocorticoid-induced hypogonadism in adult men. The potential benefits of testosterone on emotional and physical health generally outweigh potential side effects like behavioral changes, acne, body odor, rapid growth spurts, and epiphyseal closure. A recent retrospective review indicated testosterone is generally well-tolerated and perceived as beneficial by individuals with DMD and their families.
To mimic normal puberty, testosterone replacement should begin at a low dose and gradually increase to adult replacement doses over several years. Intramuscular or topical preparations can be used. Testosterone levels should be closely monitored. Consideration should be given to assessing lipids, hemoglobin, hematocrit, and blood glucose in treated individuals. If functional or cardiac status declines, discontinuing or reducing testosterone therapy should be considered.
Adrenal Insufficiency: Adrenal insufficiency due to hypothalamic-pituitary-adrenal (HPA) axis suppression is a rare but life-threatening condition that can occur if glucocorticoids are abruptly stopped due to illness or therapy discontinuation. All individuals prescribed glucocorticoids should be educated about adrenal crisis signs, symptoms, and management. They should receive prescriptions for intramuscular hydrocortisone for emergency home administration (50 mg for children <25 kg, 100 mg for children >25 kg, and 100 mg for adolescents and adults). Stress-dose glucocorticoids (double or triple the daily dose, up to 3 times per day) may be needed during severe illness, major trauma, or surgery in individuals taking more than 12 mg/m2 per day of prednisone or deflazacort. Glucocorticoid therapy should not be abruptly discontinued but tapered over weeks to months to allow HPA axis recovery. The PJ Nicholoff Steroid Protocol (Figure 3) is an appropriate approach for glucocorticoid tapering.
Gastrointestinal and Nutritional Management in Duchenne Muscular Dystrophy
Gastrointestinal and nutritional complications are common in DMD, including weight gain or loss, dietary imbalances, fluid imbalances, low bone density, swallowing dysfunction, and mandibular contractures. Contributing factors include glucocorticoid treatment, reduced energy expenditure, and immobility. These imbalances can negatively affect respiratory, skeletal muscle, and cardiac systems.
The goals of nutritional care are to prevent overweight or obesity and undernutrition or malnutrition through regular growth and weight assessments. Nutritional care also aims to promote a healthy, balanced diet with optimal intake of calories, protein, fluids, and micronutrients, especially calcium and vitamin D. Robust evidence-based nutrition research specific to DMD is lacking, so recommendations are adapted from general population guidelines. The care team should include a registered dietitian nutritionist with DMD experience, who should see the individual at every clinic visit, starting at diagnosis. More frequent dietitian monitoring is needed when weight gain or loss is anticipated. A physical therapist should be consulted to design safe exercise programs for individuals at risk of overweight. A speech-language pathologist should assess for suspected dysphagia. A gastroenterologist should be consulted for constipation, gastroesophageal reflux, gastrointestinal motility issues, and gastrostomy tube placement. Figure 5 provides an overview of recommended gastrointestinal and nutritional assessments and interventions.
Figure 5. Gastrointestinal and nutritional management algorithm for Duchenne muscular dystrophy, including dietary assessment and interventions for swallowing and digestive issues.
Nutritional Assessment and Planning: At each clinic visit, a registered dietitian nutritionist should assess nutritional status, track weight and height, and create a specific nutritional plan. Good nutritional status is defined as weight-for-length or BMI-for-age between the 10th and 85th percentiles on standard growth charts. If BMI cannot be calculated due to height measurement limitations, weight-for-age percentiles should be used. However, standard growth charts may not be optimal for DMD due to altered body composition in this population.
Patients and families should adopt healthy, balanced eating patterns as recommended in the Dietary Guidelines for Americans. Adequate fluid intake should be emphasized to prevent dehydration, which increases constipation and renal dysfunction risk. Panel 3 provides a general nutritional plan for people with DMD.
Bone health monitoring requires annual assessments of dietary calcium intake and serum 25-hydroxy-vitamin D levels. If calcium intake is below recommended levels or serum 25-hydroxy-vitamin D is less than 30 ng/mL, dietary adjustments and nutrient supplementation should follow Institute of Medicine guidelines. Bone health and osteoporosis management are detailed in part 2 of this review series.
DMD-Specific Nutritional Risks: Individuals with DMD are at risk of overweight or obesity early in life, and undernutrition or malnutrition as they approach adulthood. Early childhood glucocorticoid therapy increases overweight/obesity risk due to increased appetite, calorie intake, and sodium/fluid retention. Loss of ambulation reduces activity, lowering caloric needs and increasing overweight/obesity risk. To address these risks, a nutritional plan should include specific recommendations for calorie, protein, micronutrient, and fluid intake (Panel 3). Caloric needs are estimated by calculating resting energy expenditure and adjusting for activity level (Panel 3). Healthy eating habits, as per American Academy of Pediatrics Committee on Nutrition guidelines for obesity prevention, should be adopted by the entire family. If excessive weight gain occurs, an obesity management plan addressing diet and physical activity is needed.
Swallowing dysfunction (dysphagia) is common and often progressive in DMD. Anticipatory dysphagia assessment is important and should be done regularly. Screening questions should focus on perceived difficulty swallowing liquids and solids, food sticking in the throat, mealtime duration, and eating interference with quality of life. Affirmative responses warrant speech-language pathologist consultation for comprehensive assessment, including a video fluoroscopic swallowing study.
Unintentional weight loss often occurs before and during dysphagia onset. BMI or weight percentiles may decrease from overweight/obese to normal or underweight (malnutrition) ranges due to feeding difficulties and disease progression. Care considerations for reducing underweight/malnutrition risk during this transition are in the Appendix.
Early and ongoing gastrostomy tube feeding discussions facilitate timely intervention when clinically indicated. Gastrostomy tube placement should be viewed as a necessary and positive intervention when progressive weakness impairs self-feeding and swallowing. Indications include malnutrition unresponsive to oral intake improvement, moderate/severe dysphagia, and inadequate hydration maintenance. Gastrostomy tube feeding stabilizes or improves nutritional status in undernourished DMD individuals. Benefits should be weighed against respiratory, cardiac, and anesthetic risks of the procedure.
Common Gastrointestinal Problems: Constipation is a very frequent DMD complication. Risk factors include decreased colonic transit time, immobility, abdominal muscle weakness, and dehydration (Panel 3). Daily osmotic laxatives like polyethylene glycol, milk of magnesia, or lactulose may be needed. Retrograde enemas may help with fecal impaction.
Gastroesophageal reflux risk factors in DMD include esophageal dysmotility, delayed gastric emptying, glucocorticoid therapy, and scoliosis. Treatment includes gastric acid suppression with histamine-2 receptor antagonists (ranitidine) or proton-pump inhibitors (lansoprazole, omeprazole). Proton-pump inhibitor benefits must be balanced against potential risks like increased community-acquired pneumonia, chronic kidney disease, and bone fracture. Dietary approaches include smaller, more frequent meals and reduced dietary fat intake.
As skeletal muscle weakness progresses, delayed gastric emptying (gastroparesis) can occur, leading to postprandial abdominal pain, nausea, vomiting, early satiety, and appetite loss. Gastric emptying time can be assessed using scintigraphic gastric emptying scans. Treatment options include dietary modification, pharmacological therapy, and postpyloric feeding via gastrojejunal feeding tubes.
Conclusions and Future Directions
Part 1 of this three-part DMD care considerations update has provided guidance on diagnosis, neuromuscular, rehabilitation, endocrine, and gastrointestinal management. Key updates include care considerations for female DMD carriers, overviews of new molecular and genetic therapies, advances in rehabilitation assessments and technologies, new endocrine guidance for growth, puberty, and adrenal insufficiency, and insights into DMD-specific nutritional complications like obesity and malnutrition.
Newborn screening possibilities and emerging disease-modifying treatments emphasize the increasing importance of early treatment initiation. However, optimal timing for new therapy initiation will be crucial for newborn screening implementation decisions. Non-invasive prenatal testing for DMD is likely to become clinically available, enabling earlier detection of affected fetuses even in families without prior DMD history.
New dystrophin restorative therapies are emerging, and more data are accumulating on optimal glucocorticoid regimens. Future care considerations must address the role of new compounds in overall DMD management, especially alongside long-term glucocorticoid therapy benefits. As new therapies prove safe and effective, personalized DMD treatment approaches will become possible, tailoring therapy combinations to individual mutations. Endocrine management research needs include RCTs to better understand growth hormone and testosterone therapy risks and benefits, and to clarify optimal indications, timing, and dosing.
Rehabilitation management continues to advance with improved clinical and functional assessments across the lifespan. Technological advancements will likely drive increased use of activity monitoring combined with clinically meaningful biomarkers for therapy assessment. Robotics and other technologies will enhance independence, participation, and quality of life. Emerging dystrophin-restorative therapies may improve exercise capacity and safety. Physical therapists, occupational therapists, speech-language pathologists, and orthotists, alongside new technologies, will optimize musculoskeletal management and function.
Gastrointestinal and nutritional management research needs include resting energy expenditure (indirect calorimetry) and total energy expenditure (doubly labeled water method) assessments to better define energy needs in DMD. Specific nutritional strategies, like protein-enriched, fructose-enriched diets, branched-chain amino acid supplementation, and the influence of nutritional status on DMD outcomes (life expectancy, function, quality of life) require further investigation. DMD-specific growth charts and accurate body composition assessment techniques are needed. Understanding unique determinants of obesity in boys with DMD is crucial for developing effective prevention and management strategies, including pharmacological options. Safe and effective physical activity prescriptions could positively impact nutritional status, mobility, and social engagement throughout the lives of individuals with DMD.
Supplementary Material
Appendix with expanded tables and additional details on assessments and interventions.
Panel 1: Roles and Responsibilities of the Neuromuscular Specialist in Duchenne Muscular Dystrophy Care
- Comprehensive Disease Trajectory Assessment: Utilize validated tools to assess each patient’s unique disease progression, establishing expected clinical course, prognosis, and potential complications.
- Customized Treatment Plan Development: Select therapeutic interventions based on assessment data to create personalized treatment plans addressing individual patient and family needs and goals, optimizing outcomes and quality of life as defined by them.
- Multidisciplinary Care Coordination: Engage relevant clinicians to implement assessments, interventions, and treatment plans, ideally within a dedicated multidisciplinary DMD clinic led and coordinated by the neuromuscular specialist. Assist in the care of female carriers, including cardiac assessment.
- First-Line Medical Advisor: Serve as the primary medical advisor to patients and families, guiding them in defining and revising care goals over time, and personalizing risk-benefit analyses of therapeutic interventions. This includes technological interventions for respiratory and cardiac management, surgical and non-surgical interventions, pharmacological interventions, and clinical trial participation.
- Advocate for High-Quality Care: Champion high-quality DMD care within patient institutions and communities, addressing care transitions from pediatric to adult providers and ensuring hospital care meets patients’ unique medical, physical, and psychosocial needs.
- End-of-Life Care Navigation: Support patients and families in navigating end-of-life care, preserving comfort, dignity, and quality of life as defined by each individual patient and family.
Panel 2: Rehabilitation Assessments and Interventions Across All Disease Stages for Duchenne Muscular Dystrophy Patients
Assessment
Multidisciplinary rehabilitation assessment every 6 months, or more frequently if concerns arise, status changes occur, or specific needs are identified (refer to Appendix for detailed assessment list).
Intervention
Direct Treatment
Direct therapy provided by physical therapists, occupational therapists, and speech-language pathologists, tailored to individual needs, disease stage, therapy response, and tolerance, throughout the patient’s lifespan.
Prevention of Contracture and Deformity
- Daily Preventive Home Stretching: 4–6 times per week, regular stretching of ankles, knees, and hips, with later addition of wrist, hand, and neck stretches as indicated by assessment.
- Targeted Stretching: Stretching of structures known to be at risk of contracture and deformity, and those identified during individual assessments.*
- Orthotic Intervention, Splinting, Casting, Positioning, and Equipment:
- AFOs (Ankle-Foot Orthoses) for nighttime stretching, ideally started preventatively at a young age for better tolerance.
- AFOs for daytime stretching or positioning in non-ambulatory phases.
- Wrist or hand splints for stretching wrist and finger flexors/extensors, typically in non-ambulatory phases.
- Serial casting, applicable in both ambulatory and non-ambulatory phases.
- Passive/motorized supported standing devices, when maintaining good standing alignment becomes challenging and contractures are not severe enough to impede positioning or tolerance.
- KAFOs (Knee-Ankle-Foot Orthoses) with locked knee joints, an option for late ambulatory and non-ambulatory stages.
- Custom seating in manual and motorized wheelchairs (solid seat, back, hip guides, lateral trunk supports, adductors, headrest).
- Power positioning components on motorized wheelchairs (tilt, recline, elevating leg rests, standing support, adjustable seat height).
Exercise and Activity
Regular submaximal, aerobic activity or exercise (e.g., swimming, cycling) with assistance as needed. Avoid eccentric and high-resistance exercise. Monitor for overexertion, respect need for rest and energy conservation, and be mindful of potentially reduced cardiorespiratory exercise capacity and muscle damage risk even with good clinical function.
Falls and Fracture Prevention and Management
- Minimize fall risks in all environments.
- Physical therapist support for orthopedics in rapid team management of long-bone fractures and provision of rehabilitation to maintain ambulation and/or supported standing capabilities.
Management of Learning, Attentional, and Sensory Processing Differences
Collaborative management with the team, based on identified concerns and assessments.
Assistive Technology and Adaptive Equipment
Planning and education with assessment, prescription, training, and advocacy for funding.
Participation
Support participation in all areas of life at all disease stages.
Pain Prevention and Management
Pain prevention and comprehensive management throughout life as needed.
AFOs=ankle-foot orthoses. KAFOs=knee-ankle-foot orthoses. *Areas typically at risk of contracture and deformity include hip flexors, iliotibial bands, hamstrings, plantar flexors, plantar fascia, elbow flexors, forearm pronators, long wrist and finger flexors and extensors, lumbricals, and cervical extensors; isolated joint contracture into hip and knee flexion and plantar flexion, varus at hindfoot and forefoot, elbow flexion, wrist flexion or extension, and finger joints; and deformity of the vertebral column and chest wall including scoliosis, excessive kyphosis or lordosis, and decreased chest wall mobility.
Panel 3: General Nutritional Plan for Individuals with Duchenne Muscular Dystrophy
This general nutritional plan is adapted from recommendations for the general healthy population and is not DMD-specific. It provides methods to assess energy, protein, fluid, and micronutrient requirements based on Dietary Reference Intakes. To meet daily nutritional needs and minimize chronic disease risk, adults should consume 45–65% of total calories from carbohydrates, 20–35% from fat, and 10–35% from protein. Acceptable ranges for children are similar, with infants and younger children needing a slightly higher proportion of fat.
Overall Caloric Needs
Total caloric needs are based on total energy expenditure, which equals resting energy expenditure (REE) multiplied by a physical activity factor.
Indirect calorimetry is the most accurate REE measurement, but REE can be estimated in steroid-treated ambulatory boys with DMD (aged 10–17 years) using the Schofield weight equation: REE (kcal) = (17.7 × weight in kg + 657) × 4.182/1000. Calorie needs may decrease substantially after ambulation loss, and REE may be lower than pre-ambulation loss REE.
Physical activity factors for boys aged 3–18 years are: sedentary (1.00), low active (1.13), active (1.26), and very active (1.42).
Calculated energy/calorie intake needs adjustment for weight maintenance, loss, or gain goals.
Protein
Recommended Dietary Allowance (RDA) for protein varies by age: children aged 4–13 years: 0.95 g/kg body weight/day; ages 14–18 years: 0.85 g/kg/day; men aged 19+ years: 0.80 g/kg/day.
Fluids
Recommended fluid intake (total beverages, including water) is weight- or age-based.
Weight-based Holliday–Segar maintenance fluid method: 1–10 kg body weight: 100 mL/kg; 10–20 kg: 1000 mL + 50 mL/kg over 10 kg; >20 kg: 1500 mL + 20 mL/kg over 20 kg.
Age-based Dietary Reference Intake values for fluids: boys and girls aged 4–8 years: 1.2 L (approx. 5 cups); boys aged 9–13 years: 1.8 L (approx. 8 cups); boys aged 14–18 years: 2.6 L (approx. 11 cups); men aged 19+ years: 3.0 L (approx. 13 cups).
Micronutrients
Follow age-based RDA, except for vitamin D deficiency (25-hydroxyvitamin D <30.0 ng/mL). A multivitamin/mineral supplement is needed if calorie intake is low.
References
List of original references from the source article. (To be added based on original article’s reference list).
Contributor Information
List of contributor information from the source article. (To be added based on original article’s contributor list).
DMD Care Considerations Working Group (CCWG)
List of DMD Care Considerations Working Group members from the source article. (To be added based on original article’s working group list).
Acknowledgements
Acknowledgement section from the source article. (To be added based on original article’s acknowledgement section).
Declaration of Interests
Declaration of Interests section from the source article. (To be added based on original article’s declaration section).
Associated Data
Associated Data section from the source article. (To be added based on original article’s associated data section).