Introduction
Fabry disease (FD) is a progressive, inherited condition that can severely impact multiple organs in the body. As a lysosomal storage disorder linked to the X chromosome, FD arises from a deficiency in the enzyme α-galactosidase A (α-Gal A). This deficiency, caused by mutations in the GLA gene, leads to the buildup of a fatty substance called globotriaosylceramide (Gb3) within cells. Over time, this accumulation damages various tissues and organs, including the heart, kidneys, nervous system, and skin. Cardiac complications are particularly serious, often leading to life-threatening conditions and reduced lifespan.
Fortunately, Fabry disease is treatable. Enzyme replacement therapy (ERT) and chaperone therapy are disease-specific treatments that can significantly alter the course of the disease. Because effective treatments are available, early and accurate Fabry Disease Diagnosis is paramount. Timely diagnosis allows for the initiation of treatment, which can help to reduce morbidity and mortality associated with FD. This article provides an updated overview of fabry disease diagnosis, highlighting key diagnostic strategies and screening approaches for healthcare professionals.
Understanding Fabry Disease: Epidemiology and Pathogenesis
The estimated prevalence of Fabry disease ranges from 1 in 40,000 to 1 in 117,000 individuals. However, newborn screening programs suggest that the actual prevalence may be higher. For instance, a newborn screening study in Spain found a prevalence of FD in males as high as 1 in 7,575. Studies focusing on high-risk populations have also revealed a higher prevalence of GLA gene variants.
Fabry disease is caused by mutations in the GLA gene, which is located on the X chromosome. These mutations result in a deficiency or absence of α-Gal A activity. This enzyme is crucial for breaking down Gb3. When α-Gal A is deficient, Gb3 accumulates within cells throughout the body, causing a range of symptoms and organ damage.
While FD is X-linked, females, who have two X chromosomes, can also be affected. Although often manifesting later and sometimes less severely than in males, females can experience significant clinical symptoms. The presentation of Fabry disease varies widely, even within the same family, ranging from severe, early-onset classic forms to milder, later-onset variants.
Two primary phenotypes of Fabry disease are recognized: classic and variant. Males with classic FD typically develop symptoms in childhood or adolescence. These may include episodes of severe pain in the extremities (acroparesthesias), neuropathic pain, small, dark red spots on the skin called angiokeratomas, sweating abnormalities, gastrointestinal issues, and eye problems like corneal opacities (cornea verticillata). In adults, vascular complications, kidney failure, unexplained left ventricular hypertrophy (LVH), and neurological symptoms such as stroke or hearing loss can emerge.
Variant FD often presents later in life with primarily single-organ involvement, such as kidney, heart, or brain. Females exhibit a broad spectrum of symptoms, ranging from being asymptomatic to severely affected, generally with symptom onset occurring later than in males.
Globotriaosysphingosine (lyso-Gb3), a deacylated metabolite of Gb3, is also implicated in the disease process. Both Gb3 and lyso-Gb3 deposition contribute to cellular dysfunction, leading to fibrosis, inflammation, and organ damage. Plasma lyso-Gb3 levels have been shown to correlate with disease severity and may decrease with effective treatment.
Diagnostic Approaches for Fabry Disease
Diagnosing classic Fabry disease in males can be relatively straightforward, but fabry disease diagnosis in females and individuals with variant forms can be more challenging. A comprehensive diagnostic approach is essential, integrating clinical evaluation, biochemical testing, genetic analysis, and imaging studies.
A detailed patient history, including family history, is crucial. Physical examination may reveal characteristic signs like angiokeratomas or cornea verticillata, which are highly suggestive of FD.
Initial biochemical testing in males suspected of having FD should include measurement of α-Gal A enzyme activity. Significantly reduced α-Gal A activity in males strongly indicates Fabry disease. However, enzyme activity levels can be variable, particularly in females, and may sometimes fall within the normal range despite the presence of the disease.
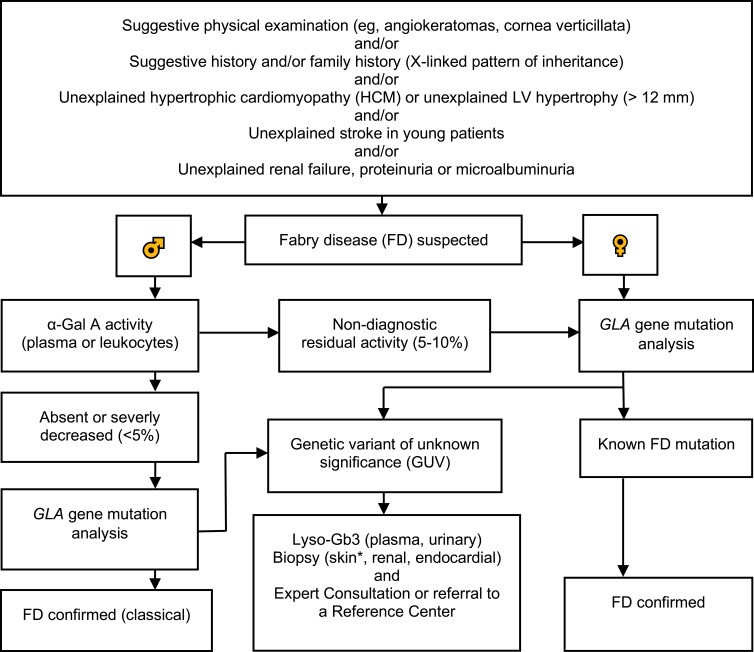 Diagnostic algorithm for Fabry disease, showing steps for males and females including enzyme assay, genetic testing, and biomarker analysis.
Diagnostic algorithm for Fabry disease, showing steps for males and females including enzyme assay, genetic testing, and biomarker analysis.
Genetic testing to confirm mutations in the GLA gene is mandatory for both males and females to definitively establish fabry disease diagnosis. GLA gene mutations can be associated with classic FD, variant phenotypes, or variants of unknown significance (GVUS).
Figure 1 presents an updated diagnostic algorithm for Fabry disease. For males, initial screening often involves measuring α-Gal A activity. If activity is deficient, genetic testing is recommended. In cases of borderline enzyme activity (5–10%), genetic testing is crucial for confirmation. For females, genetic testing is often the primary diagnostic step due to the variability of enzyme activity.
For individuals with GVUS, further investigation is needed to determine the clinical significance of the variant. Lyso-Gb3 measurement can be particularly helpful in these cases, as elevated lyso-Gb3 levels can support a diagnosis of Fabry disease even when genetic variants are of uncertain significance.
Furthermore, for patients diagnosed with Fabry disease, assessing amenability to chaperone therapy with migalastat can be performed using a validated in vitro assay. This assay determines if a patient’s specific GLA mutation is responsive to migalastat.
The Role of Biomarkers in Fabry Disease Diagnosis
Biomarkers play an increasingly important role in fabry disease diagnosis and management. Lyso-Gb3 has emerged as a significant biomarker, especially in cases with variable α-Gal A activity or GVUS. Lyso-Gb3 levels can help to differentiate between classic and variant phenotypes and may even identify individuals with FD before clinical symptoms are apparent. Its particular utility lies in situations with phenotypic variability, most notably in females.
In the context of cardiac involvement, high-sensitive Troponin T (hsTNT) shows promise as a biomarker. Elevated hsTNT levels may indicate myocardial damage in Fabry cardiomyopathy and correlate with the severity of cardiac involvement detected by cardiac magnetic resonance imaging (CMR).
While NT-proBNP levels can be elevated in Fabry patients with cardiac involvement, they do not appear to correlate well with myocardial fibrosis or disease progression. Therefore, hsTNT and lyso-Gb3 are currently considered more valuable biomarkers for Fabry disease diagnosis and monitoring.
Diagnosing Organ-Specific Involvement
Following a confirmed fabry disease diagnosis, assessing the extent of organ involvement is critical for guiding management and treatment decisions.
Cardiac Involvement
Echocardiography is typically the initial step in evaluating cardiac involvement in Fabry disease. Characteristic echocardiographic findings include concentric LVH and a prominent papillary muscle. Diastolic dysfunction is also commonly observed. However, left ventricular ejection fraction (EF) is usually preserved until late stages of the disease.
For a more comprehensive cardiac assessment, cardiac magnetic resonance imaging (CMR) is recommended. Late gadolinium enhancement CMR (LGE-CMR) is the gold standard for detecting myocardial fibrosis, a key indicator of Fabry cardiomyopathy. In patients who cannot undergo CMR, speckle-tracking echocardiography (STE) can provide indirect evidence of cardiac involvement.
T1-mapping on cardiac MRI can further aid in differentiating Fabry cardiomyopathy from other causes of LVH. Abnormal native myocardial T1 values are highly prevalent in FD patients with LVH. Furthermore, PET-MR imaging is being investigated for the early detection of cardiac involvement, even before hypertrophy develops, by identifying myocardial inflammation.
Renal Involvement
Initial assessment of renal function in Fabry disease includes standard tests such as serum creatinine, estimated glomerular filtration rate (GFR), and urine protein analysis. Spot urine albumin-to-creatinine ratio is highly recommended for early detection of microalbuminuria. Cystatin C-based GFR calculation is also recommended as cystatin C is a more sensitive marker for early renal dysfunction than creatinine. Kidney ultrasound should also be performed.
While urine lyso-Gb3 has not been shown to be a reliable marker for renal involvement, proteinuria and albuminuria are crucial indicators of early kidney damage in FD. Persistent albuminuria or proteinuria warrants a renal biopsy to evaluate the extent of Fabry nephropathy.
Neurologic Involvement
Diagnosing neurological involvement in Fabry disease currently relies on clinical evaluation and imaging studies, as specific blood biomarkers are lacking. Neurological symptoms such as pain (paresthesias, dysesthesias, chronic burning pain), sweating abnormalities, hearing loss, and gastrointestinal issues should prompt further neurological investigation.
Brain MRI is a key diagnostic tool, often revealing characteristic white matter hyperintensities, dolichoectasia (dilation of blood vessels), and infarcts in patients with classic FD. Audiometry is used to assess hearing loss, another potential neurological manifestation.
Screening Strategies for Fabry Disease
Screening for Fabry disease aims to identify asymptomatic individuals who are likely to have the condition, allowing for earlier diagnosis and treatment. However, general population newborn screening for FD is not yet widely implemented due to various factors, including cost-effectiveness considerations.
Currently, screening for Fabry disease is primarily recommended in high-risk populations and families of individuals diagnosed with FD. For males, biomarker-based screening using α-Gal A activity or lyso-Gb3 levels is effective. Enzyme-based screening is less reliable in females due to the variability of α-Gal A activity. Retinal vessel diameter assessment may also be a helpful screening tool.
High-Risk Population Screening
Screening specific high-risk populations can significantly increase the detection rate of Fabry disease. These high-risk groups include:
- Patients with unexplained left ventricular hypertrophy (LVH) or hypertrophic cardiomyopathy (HCM): Studies have shown a notable prevalence of FD in individuals with unexplained LVH.
- Patients with end-stage renal disease, unexplained proteinuria, or microalbuminuria: Fabry disease should be considered in patients undergoing hemodialysis, kidney transplantation, or those with unexplained renal findings.
- Individuals (aged 15–55 years) with unexplained stroke: Particularly in younger individuals, Fabry disease is a potential cause of stroke.
Screening these high-risk groups using α-Gal A activity measurement, lyso-Gb3 levels, and genetic testing can lead to earlier fabry disease diagnosis and intervention.
Family Screening
Family screening is crucial in Fabry disease due to its inherited nature. Pedigree analysis and genetic counseling are essential for families affected by FD. Relatives of diagnosed individuals should be offered genetic testing to identify those who may also have inherited the condition, even if they are currently asymptomatic.
Conclusion
Fabry disease is a serious, progressive, multi-system disorder that can significantly reduce life expectancy. Early fabry disease diagnosis is critical for improving patient outcomes. Utilizing updated diagnostic algorithms, incorporating biomarkers like lyso-Gb3 and hsTNT, and implementing targeted screening strategies in high-risk populations are essential steps in achieving timely diagnosis and initiating appropriate management, including enzyme replacement therapy or chaperone therapy. By focusing on early detection and comprehensive diagnostic approaches, healthcare professionals can make a significant difference in the lives of individuals affected by Fabry disease.
References
1–4
5
6,7
8
9
10
11–14
15–19
20,21
22
23,24
25
26
27,28
29–31
30,32–34
30,35
30,34–36
37
38
39
5,40
41
10,42,43
10
44
45,46
27,47
10
10
10,27,31,43,48,49
50,51
52
37,48
48
42
53,54
53,54
55
55
56,57
42,55
58
59,60
58,59
61
58
62
62
63
62,64
64,65
53,66
27
67
63
65
68,69
70
69
71
72
73–75
76,77
78
78
78
11,79
9
49,80
19
81
82
20,23
80
15,16,18,19
10
43