Introduction
Hereditary transthyretin amyloidosis (hATTR) is a progressive, inherited disorder characterized by the accumulation of misfolded transthyretin (TTR) protein in various organs and tissues throughout the body. This rare condition, stemming from mutations in the TTR gene, presents a significant Hattr Amyloidosis Diagnosis challenge for clinicians due to its heterogeneous clinical manifestations and broad spectrum of organ involvement. While polyneuropathy is a hallmark feature, hATTR amyloidosis can affect the heart, eyes, kidneys, and gastrointestinal system, leading to a complex clinical picture that often mimics other, more common diseases. The insidious onset and varied presentation of hATTR amyloidosis frequently result in delayed or missed diagnoses, hindering timely access to disease-modifying therapies that can significantly alter the course of this debilitating condition. Therefore, a heightened awareness of the diverse clinical phenotypes of hATTR amyloidosis, coupled with advancements in diagnostic tools and strategies, is crucial for improving early hattr amyloidosis diagnosis and ultimately enhancing patient outcomes. This review delves into the multifaceted aspects of hattr amyloidosis diagnosis, highlighting the complexities, common pitfalls, and evolving approaches to facilitate accurate and prompt identification of this challenging disease.
Epidemiology and Genetic Basis of hATTR Amyloidosis
Historically considered a geographically localized disease, hATTR amyloidosis is now recognized as a global entity, with cases reported across at least 29 countries. While certain regions, like Portugal, Sweden, and Japan, exhibit higher prevalence due to founder mutations, the increasing availability of genetic testing and growing clinical awareness have led to the identification of hATTR amyloidosis in non-endemic areas as well. The underlying cause of hATTR amyloidosis lies in mutations of the TTR gene, located on chromosome 18. Over 120 pathogenic variants have been identified, with the p.Val30Met mutation being the most prevalent worldwide. This genetic heterogeneity contributes to the diverse clinical presentations observed in hATTR amyloidosis, further complicating the hattr amyloidosis diagnosis process. Inherited in an autosomal dominant manner, hATTR amyloidosis exhibits variable penetrance, meaning not everyone carrying a mutation will develop the disease, and the age of onset and disease phenotype can vary even within families. This variability is influenced by genetic modifiers, geographic origin, and even parental inheritance patterns, adding layers of complexity to both disease manifestation and hattr amyloidosis diagnosis.
Pathophysiology: Understanding the Roots of hATTR Amyloidosis
At its core, hATTR amyloidosis is a protein misfolding disorder. Transthyretin, normally a stable tetramer responsible for transporting thyroxine and retinol-binding protein, becomes unstable due to TTR gene mutations. These mutations promote the dissociation of the tetramer into monomers, which misfold and aggregate into amyloid fibrils. These amyloid fibrils deposit extracellularly in various organs, disrupting their normal function. The insidious nature of amyloid deposition, often starting years before clinical symptoms emerge, underscores the importance of early hattr amyloidosis diagnosis before irreversible organ damage occurs. It’s crucial to differentiate hATTR amyloidosis from wild-type transthyretin amyloidosis (ATTRwt), an age-related condition where normal TTR protein misfolds and deposits, primarily in the heart. While both forms result in amyloid deposition, the underlying mechanisms and genetic basis are distinct, impacting diagnostic approaches and therapeutic strategies.
Clinical Features: A Spectrum of Manifestations
The clinical presentation of hATTR amyloidosis is remarkably heterogeneous, ranging from predominantly neuropathic to primarily cardiac phenotypes, and often a combination of both. This clinical variability is a major factor contributing to the challenges in hattr amyloidosis diagnosis.
Neurological Manifestations
Polyneuropathy is a common and often initial manifestation of hATTR amyloidosis. Early-onset hATTR-Val30Met amyloidosis, typically seen in endemic regions, often presents with a length-dependent, small-fiber neuropathy. Patients may experience neuropathic pain, numbness, and temperature insensitivity in the feet, progressing upwards. Autonomic neuropathy is frequently associated, leading to gastrointestinal issues (diarrhea, constipation, nausea, early satiety), orthostatic hypotension, and genitourinary dysfunction.
Late-onset hATTR amyloidosis, including non-Val30Met variants and late-onset Val30Met cases, exhibits a more variable neurological presentation. Symptoms can start distally or proximally, affecting lower and upper limbs, sometimes mimicking carpal tunnel syndrome. Motor involvement can be more prominent in late-onset cases compared to early-onset.
Cardiac Involvement
Cardiac amyloidosis is a significant aspect of hATTR amyloidosis and can be a predominant feature in certain TTR mutations (e.g., Val122Ile, Ile68Leu, Thr60Ala). Amyloid infiltration of the heart leads to progressive ventricular wall thickening, diastolic dysfunction, conduction abnormalities, and heart failure with preserved ejection fraction. Cardiac arrhythmias and conduction blocks are common and pose a risk of sudden cardiac death, highlighting the importance of cardiac evaluation in hattr amyloidosis diagnosis.
Other Organ Involvement
Beyond the nervous system and heart, hATTR amyloidosis can affect other organs:
- Ocular: Ocular manifestations are frequent and increase with disease duration. Dry eye syndrome, amyloid deposits in the iris or lens capsule, glaucoma, vitreous opacities, and retinal angiopathy are among the reported ocular findings.
- Renal: Kidney involvement can manifest as nephrotic syndrome and progressive renal failure, although it’s more common in specific mutations like Val30Met, particularly in Portuguese patients.
- Central Nervous System (CNS): While less frequent, CNS involvement due to leptomeningeal amyloid angiopathy can occur, leading to stroke, hemorrhage, hydrocephalus, ataxia, spasticity, seizures, hearing loss, and dementia.
The multisystem nature and diverse clinical presentations of hATTR amyloidosis necessitate a comprehensive diagnostic approach to accurately identify the condition and differentiate it from other mimicking disorders.
The Diagnostic Journey of hATTR Amyloidosis
Given the clinical heterogeneity of hATTR amyloidosis, achieving a timely and accurate hattr amyloidosis diagnosis requires a multi-step process. This journey often begins with clinical suspicion, followed by tissue biopsy and genetic testing for confirmation.
Clinical Suspicion and Initial Assessment
Raising clinical suspicion is the first and often most crucial step in hattr amyloidosis diagnosis. Physicians should consider hATTR amyloidosis in patients presenting with:
- Unexplained progressive peripheral neuropathy, especially with small fiber involvement or autonomic dysfunction.
- Cardiomyopathy, particularly hypertrophic cardiomyopathy with preserved ejection fraction, especially if accompanied by neuropathy.
- Multisystem symptoms involving the nervous system, heart, eyes, kidneys, or gastrointestinal tract.
- Family history of amyloidosis or unexplained neuropathy or cardiomyopathy.
In non-endemic areas, where familial history might be less apparent, a higher index of suspicion is essential. The diagnostic delay in these regions is often longer due to the rarity of the disease and its mimicry of more common conditions. Initial assessment should include a thorough neurological examination, cardiac evaluation (ECG, echocardiogram), and assessment for autonomic dysfunction.
Tissue Biopsy and Amyloid Detection
Once clinical suspicion is raised, tissue biopsy is a critical step to confirm amyloid deposition. Biopsy can be performed on various tissues, including:
- Nerve: Sural nerve biopsy has a sensitivity of around 80% for detecting amyloid in hATTR-PN.
- Fat pad: Abdominal fat aspiration is a less invasive option with variable sensitivity (14-83%).
- Labial salivary gland: Salivary gland biopsy is another minimally invasive procedure, particularly useful in unexplained axonal neuropathy cases.
- Myocardium: Cardiac biopsy is more invasive but may be necessary in cases with predominant cardiac involvement, especially to differentiate between hATTR-CM and ATTRwt or other cardiomyopathies.
- Skin, kidney, gastrointestinal mucosa: These tissues can also be biopsied depending on clinical presentation.
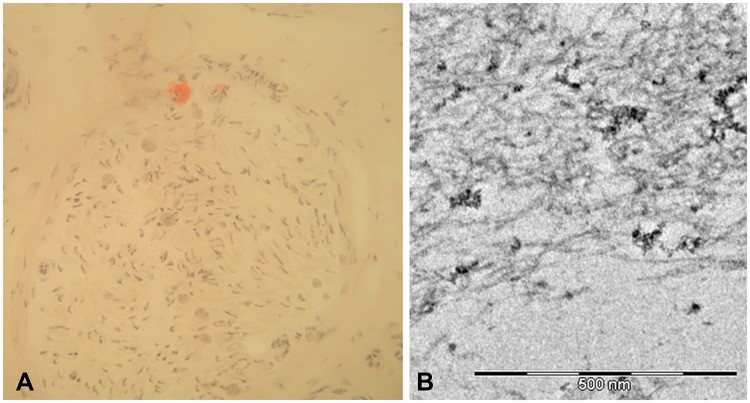
Histopathological examination of the biopsy sample with Congo red staining is the gold standard for amyloid detection. Amyloid deposits exhibit characteristic apple-green birefringence under polarized light after Congo red staining. Once amyloid is confirmed, determining the type of amyloid protein is crucial. Immunohistochemistry or mass spectrometry can identify TTR as the precursor protein, confirming the diagnosis of transthyretin amyloidosis. However, it’s important to note that amyloid deposition can be patchy, and a negative biopsy does not rule out hATTR amyloidosis, especially if clinical suspicion remains high. In such cases, repeat biopsies or proceeding to genetic testing is warranted for definitive hattr amyloidosis diagnosis.
Genetic Testing: The Gold Standard for hATTR Amyloidosis Diagnosis
TTR gene sequencing is now considered the gold standard for confirming hattr amyloidosis diagnosis, especially when clinical suspicion is high, or biopsy results are inconclusive. Sanger sequencing is the most widely used method for detecting TTR gene mutations. Genetic testing is particularly crucial in:
- Patients with suspected hATTR amyloidosis, even with negative or inconclusive biopsies.
- Individuals with a family history of hATTR amyloidosis for presymptomatic diagnosis and genetic counseling.
- Atypical presentations where clinical features mimic other conditions.
Identifying a pathogenic TTR mutation confirms the diagnosis of hATTR amyloidosis and helps in genetic counseling and family screening. With the increasing availability and affordability of genetic testing, it should be considered a primary diagnostic tool in suspected cases of hATTR amyloidosis.
Differential Diagnosis: Distinguishing hATTR Amyloidosis from Mimicking Conditions
The heterogeneous clinical presentation of hATTR amyloidosis often leads to misdiagnosis. Differential diagnosis is crucial to exclude other conditions that mimic hATTR amyloidosis. Common misdiagnoses include:
- Peripheral Neuropathies: Idiopathic axonal polyneuropathy, chronic inflammatory demyelinating polyradiculoneuropathy (CIDP), diabetic neuropathy, Charcot-Marie-Tooth disease, vasculitic neuropathy, toxic neuropathy, alcoholic neuropathy, paraproteinemic neuropathy.
- Carpal Tunnel Syndrome: Upper-limb onset hATTR amyloidosis can mimic carpal tunnel syndrome.
- Motor Neuron Disease: Motor neuropathy presentations can be mistaken for amyotrophic lateral sclerosis (ALS) or other motor neuron diseases.
- Cardiomyopathies: Hypertrophic cardiomyopathy due to sarcomeric mutations, Anderson-Fabry disease, mitochondrial cardiomyopathies, Danon disease, Noonan syndrome, wild-type ATTR amyloidosis (ATTRwt).
- Other Amyloidosis Types: Immunoglobulin light chain amyloidosis (AL amyloidosis).
Careful clinical evaluation, considering multisystem involvement, and utilizing appropriate diagnostic tests are essential to differentiate hATTR amyloidosis from these mimicking conditions and ensure accurate hattr amyloidosis diagnosis.
Advanced Diagnostic Investigations to Assess Organ Involvement
Once hATTR amyloidosis is diagnosed, further investigations are necessary to assess the extent and severity of organ involvement. These include:
- Neurological Assessments:
- Nerve Conduction Studies: To evaluate the presence and severity of peripheral neuropathy.
- Skin Biopsy: Gold standard for small fiber neuropathy diagnosis.
- Sudomotor Function Testing (e.g., Electrochemical Skin Conductance): To assess autonomic neuropathy.
- Heart Rate Variability and Orthostatic Hypotension Testing: Further autonomic function evaluation.
- Cardiac Assessments:
- Echocardiogram: To detect hypertrophic cardiomyopathy and diastolic dysfunction.
- 24-hour Holter Monitoring: To identify arrhythmias and conduction blocks.
- Echocardiography with Strain Imaging: More sensitive detection of cardiac dysfunction.
- Cardiac Magnetic Resonance Imaging (MRI): To characterize amyloid infiltration and assess myocardial fibrosis.
- Cardiac Scintigraphy (Bone Tracers – DPD, HMDP, PYP): Non-invasive imaging to detect cardiac amyloid deposition, helpful in differentiating ATTR amyloidosis from AL amyloidosis.
- Serum Biomarkers (BNP, NT-proBNP, Troponins): Prognostic markers and indicators of cardiac stress and damage.
- Ophthalmological Assessment: Comprehensive eye exam including visual acuity, intraocular pressure, Schirmer test, fundus examination, and slit-lamp examination to detect ocular manifestations.
- Renal Evaluation: Serum creatinine, proteinuria, microalbuminuria, estimated glomerular filtration rate (eGFR), and potentially serum cystatin C to assess renal function.
- Nutritional Status Assessment: Modified Body Mass Index (mBMI) to account for hypalbuminemia and assess nutritional status, particularly in patients with gastrointestinal involvement.
These comprehensive investigations provide a detailed picture of organ involvement, guiding disease staging, prognosis, and treatment decisions in hATTR amyloidosis.
Outcome Measures in hATTR Amyloidosis
Monitoring disease progression and treatment response in hATTR-PN requires the use of validated outcome measures. Commonly used tools include:
- FAP Staging System and Polyneuropathy Disability (PND) Score: Overall disease severity assessment.
- Neuropathy Impairment Score (NIS) and modified NIS+7 (mNIS+7): Quantify neuropathic impairment, including sensory, motor, and electrophysiological assessments. mNIS+7 variants (mNIS+7Alnylam, mNIS+7Ionis) are specifically designed for clinical trials in hATTR amyloidosis.
- Composite Autonomic Symptom Scale-31 (COMPASS-31) and Compound Autonomic Dysfunction Test (CADT) questionnaire: Assess autonomic symptoms.
- Rasch-built Overall Disability Scale (R-ODS): Evaluate activities of daily living.
- Norfolk Quality of Life-Diabetic Neuropathy (QoL-DN) questionnaire: Assess quality of life.
- Motor Function Tests: 6-minute walk test, 10-meter walk test, handgrip strength test.
- Sudoscan: Instrumental assessment of sudomotor function for disease monitoring.
The selection of appropriate outcome measures depends on the clinical context, research setting, and specific aspects of disease progression being monitored.
Therapeutic Implications of Early and Accurate hATTR Amyloidosis Diagnosis
The advent of disease-modifying therapies for hATTR amyloidosis has transformed the management of this once devastating disease. Liver transplantation, TTR stabilizers (tafamidis, diflunisal), and gene-silencing therapies (patisiran, inotersen) are now available treatment options. These therapies aim to reduce TTR production, stabilize TTR tetramers, or inhibit amyloid fibril formation, thereby slowing disease progression and improving patient outcomes.
However, the effectiveness of these therapies is often greater when initiated early in the disease course, before significant irreversible organ damage has occurred. Therefore, timely and accurate hattr amyloidosis diagnosis is paramount to ensure patients benefit maximally from these therapeutic advancements. Early diagnosis allows for prompt initiation of treatment, potentially delaying disease progression, preserving neurological and cardiac function, and improving overall survival. Systematic monitoring of asymptomatic TTR mutation carriers is also crucial for early detection of disease onset and timely treatment initiation.
Image alt text: Diagram illustrating therapeutic strategies for hATTR with polyneuropathy, including liver transplantation, TTR stabilizers, gene silencing therapies (RNA interference and antisense oligonucleotides), and emerging approaches, emphasizing the range of treatment options following hATTR amyloidosis diagnosis.
Conclusion: Advancing hATTR Amyloidosis Diagnosis for Improved Patient Care
Hattr amyloidosis diagnosis remains a complex and evolving field. The multisystem nature of the disease, its heterogeneous clinical presentations, and mimicry of other conditions pose significant diagnostic challenges. However, increased clinical awareness, advancements in diagnostic tools – including genetic testing, tissue biopsy techniques, and cardiac imaging – are improving the ability to accurately and promptly diagnose hATTR amyloidosis. Genetic testing has become a cornerstone of hattr amyloidosis diagnosis, especially in non-endemic regions and for familial screening. Early and accurate hattr amyloidosis diagnosis is crucial for enabling timely initiation of disease-modifying therapies, which can significantly alter the natural history of this severe disease. Continued research into novel diagnostic biomarkers and improved diagnostic algorithms are essential to further reduce diagnostic delays and ensure optimal management and outcomes for individuals affected by hATTR amyloidosis. The future of hattr amyloidosis diagnosis lies in a multidisciplinary approach, integrating clinical expertise, advanced diagnostic technologies, and a heightened awareness of this rare but treatable condition.
Abbreviations
9mTc-DPD, technetium-99m radiolabeled 2,3-dicarboxypropane-1,1-diphosphonate; 99mTc-HMDP, technetium-99m radiolabeled hydroxymethylene diphosphonate; 99mTc-PYP, technetium-99m radiolabeled pyrophosphate; ASO, antisense oligonucleotide; ATTRwt, wild-type transthyretin amyloidosis; BNP, brain natriuretic peptide; CADT, Compound Autonomic Dysfunction Test; CNS, central nervous system; COMPASS-31, Composite Autonomic Symptom Scale-31; CSF, cerebrospinal fluid; EGCG, epigallocatechin-3-gallate; eGFR, estimated glomerular filtration rate; EMA, European Medicines Agency; FAC, familial amyloid cardiomyopathy; FAP, familial amyloid polyneuropathy; FDA, Food and Drug Administration; GalNAc, N-acetylgalactosamine; hATTR, hereditary transthyretin amyloidosis; hATTR-CM, hereditary transthyretin amyloidosis with cardiomyopathy; hATTR-PN, hereditary transthyretin amyloidosis with polyneuropathy; LT, liver transplantation; mBMI, modified body mass index; mNIS+7, modified Neuropathy Impairment Score +7; MRI, magnetic resonance imaging; NIS, Neuropathy Impairment Score; NIS+7, Neuropathy Impairment Score +7; NIS-LL, Neuropathy Impairment Score-Lower Limbs; Norfolk QoL-DN, Norfolk Quality of Life-Diabetic Neuropathy; NSAID, nonsteroidal anti-inflammatory drug; NT-proBNP, N-terminal prohormone of brain natriuretic peptide; PND, Polyneuropathy Disability score; QoL, quality of life; RNAi, RNA interference; RNase H1, ribonuclease H1; R-ODS, Rasch-built Overall Disability Scale; siRNAs, small interfering RNAs; T4, thyroxine; TTR, transthyretin.
Disclosure
Dr Luigetti received financial grants (honoraria and speaking) from Akcea, Alnylam and Pfizer, and travel grants from Pfizer, Kedrion and Grifols; Dr Bisogni received financial grants (honoraria and speaking) from Alnylam, and travel grants from Pfizer, and Grifols; Dr Romano received travel grants from Akcea and Pfizer; Dr Di Paolantonio received travel grants from Akcea and Pfizer; Dr Sabatelli received financial grants (honoraria and speaking) from Akcea. The authors report no other conflicts of interest in this work.