Hereditary transthyretin amyloidosis (hATTR), previously known as Familial Amyloid Polyneuropathy, presents a significant diagnostic challenge in the field of neurology. This rare genetic disorder stems from mutations in the transthyretin (TTR) gene, leading to the systemic accumulation of amyloid deposits and subsequent dysfunction across multiple organs and tissues. While polyneuropathy is a common presenting symptom, the diverse clinical manifestations and widespread organ involvement in hATTR amyloidosis necessitate a high degree of clinical suspicion and a multifaceted approach to diagnosis. Recognizing the frequent yet often latent cardiac, ocular, and renal complications is crucial for clinicians. Notably, hypertrophic cardiomyopathy, even in its early stages, is identifiable in a substantial proportion, at least 50%, of hATTR patients. The advent of therapies targeting various stages of TTR production, including synthesis inhibition (liver transplantation and gene-silencing drugs) and TTR stabilization, has revolutionized the management of hATTR, underscoring the critical importance of timely and accurate Hattr Diagnosis to improve patient survival across different disease stages.
A QR code for video abstract about hereditary transthyretin amyloidosis (hATTR) diagnosis and clinical overview, facilitating quick access to multimedia information for medical professionals.
Epidemiology of hATTR Amyloidosis and Diagnostic Implications
Historically considered a rare condition, hATTR amyloidosis was initially recognized in major endemic clusters in Portugal, Sweden, and Japan. Subsequently, smaller endemic regions were identified in Cyprus and Majorca. Within Europe, the occurrence of hATTR amyloidosis exhibits considerable variability, with sporadic cases predominating.
However, contemporary medical practice witnesses an increasing awareness of hATTR amyloidosis among healthcare professionals, coupled with the widespread availability of advanced genetic testing. This paradigm shift suggests a likely rise in the reported incidence of hATTR amyloidosis, particularly in regions previously deemed non-endemic. Consistent with this trend, hATTR has now been documented in at least 29 countries globally, highlighting the expanding geographical understanding and hATTR diagnosis reach of this disease. This broader recognition is essential for improving global diagnostic strategies.
Genetic Basis of hATTR and its Role in Diagnosis
The TTR gene, comprising four exons, is located on chromosome 18. To date, over 120 TTR variants have been identified, with a significant majority classified as pathogenic. The most frequently encountered pathogenic variant is a point mutation leading to the substitution of valine with methionine at position 30 within the mature protein. This Val30Met mutation is a primary cause of hATTR amyloidosis in endemic regions and remains the most prevalent amyloidogenic mutation worldwide, accounting for approximately 50% of TTR variants globally. Genetic screening for this and other mutations is a cornerstone of hATTR diagnosis.
hATTR amyloidosis follows an autosomal dominant inheritance pattern. Interestingly, cases of homozygous pathogenic mutations and compound heterozygous patients have also been reported, expanding the genetic complexity of this condition.
The penetrance of hATTR can vary significantly across geographic regions. Complete penetrance has been frequently observed in multi-generational pedigrees from endemic areas. Conversely, in non-endemic regions, incomplete penetrance is more common, progressively increasing with age and approaching nearly 100% by the ninth decade of life. This variable penetrance impacts hATTR diagnosis strategies, especially in sporadic cases and non-endemic populations.
Factors influencing the age of onset and phenotypic expression in families carrying Val30Met mutations remain largely unknown. Both genetic and non-genetic modifying factors, including genotype, geographic origin, and the parent of origin of the mutation, can contribute to the variability observed in disease onset and phenotypic presentation. Understanding these genetic nuances is crucial for refining hATTR diagnosis and risk assessment.
Pathophysiology of hATTR: Implications for Understanding Disease Mechanisms and Diagnosis
Transthyretin (TTR), initially known as prealbumin, is a 55-kDa protein responsible for transporting thyroxine (T4) and retinol-binding protein. In healthy individuals, TTR circulates in a soluble form in both serum and cerebrospinal fluid (CSF). The liver is the primary site of TTR synthesis, with minor production occurring in the choroid plexus, retinal pigment epithelium, and ciliary pigment epithelia. Under normal physiological conditions, TTR exists as a homotetramer with a central channel containing two T4-binding sites, although only one site is typically occupied. Understanding the normal function and structure of TTR is foundational for comprehending the pathological processes in hATTR and improving hATTR diagnosis.
hATTR amyloidosis is categorized as a protein misfolding disease. Most pathogenic mutations destabilize the TTR tetramer compared to the wild-type protein, promoting its dissociation into monomers. This monomerization leads to misfolding, aggregation, and subsequent extracellular deposition of TTR amyloid fibrils in various organs, resulting in progressive multisystem dysfunction. Amyloid deposition is a gradual process, often starting years before the onset of clinical symptoms, similar to other neurodegenerative conditions such as Alzheimer’s and Parkinson’s disease. Early hATTR diagnosis aims to detect these changes before irreversible damage occurs.
TTR amyloid fibrils can also originate from the deposition of wild-type transthyretin protein in ATTRwt amyloidosis. This condition, also known as senile amyloidosis, is age-related and involves the structural dissociation and misassembly of wild-type TTR into amyloid fibrils. Differentiating between hATTR and ATTRwt is important for accurate hATTR diagnosis and management.
Clinical Features of hATTR Amyloidosis: Navigating Diagnostic Variability
The clinical presentation of hATTR amyloidosis is remarkably heterogeneous, ranging from a predominantly neurological phenotype in endemic cohorts to a strictly cardiological presentation in sporadic cases. This clinical variability significantly complicates hATTR diagnosis.
Table 1. Clinical Manifestations and Epidemiology of the Most Common hATTR Mutations
| Mutation | Epidemiology | Peripheral Neuropathy | Autonomic Neuropathy | Cardiomyopathy | Ocular Involvement | Gastrointestinal Involvement | Renal Involvement |
|---|---|---|---|---|---|---|---|
| Val30Met(early onset) | Portugal, Brazil | ++ | +++ | ± | + | ++ | + |
| Val30Met(late onset) | Japan, Sweden, USA,Italy, France | +++ | + | ++ | + | + | ± |
| Val122Ile | USA | ± | ± | +++ | ± | ± | ± |
| Thr60Ala | UK, USA | + | + | +++ | ± | ± | ± |
| Glu89Gln | Italy, Bulgaria | ++ | ++ | ++ | ± | + | ± |
| Ser50Arg | Japan, France, Italy, USA | +++ | +++ | ± | ± | + | ± |
| Phe64Leu | USA, Italy | ++ | ++ | ++ | ± | + | ± |
| Ile68Leu | Germany, Italy | ± | ± | +++ | ± | ± | ± |
| Ser77Tyr | USA, France, Israel | ++ | ++ | ++ | ± | + | + |
| Ile107Val | USA, France, Brazil | ++ | ++ | ++ | ± | ± | ± |
| Asp38Ala | Japan | ++ | ++ | ++ | ± | ± | ± |

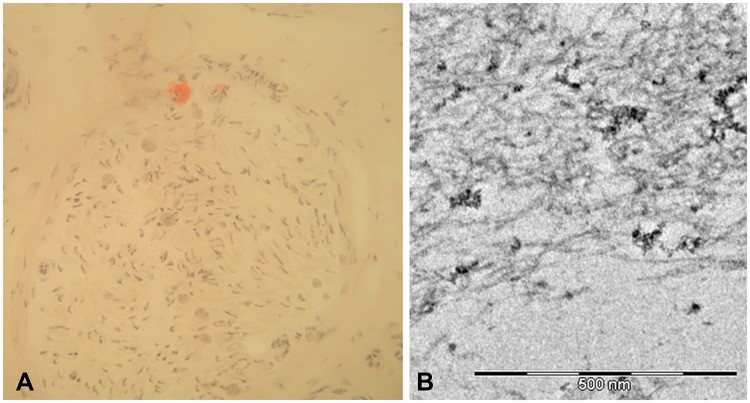
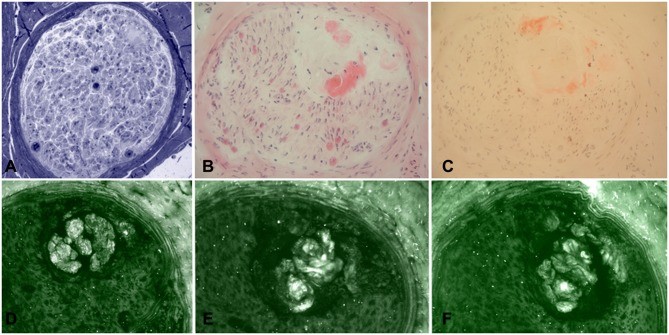
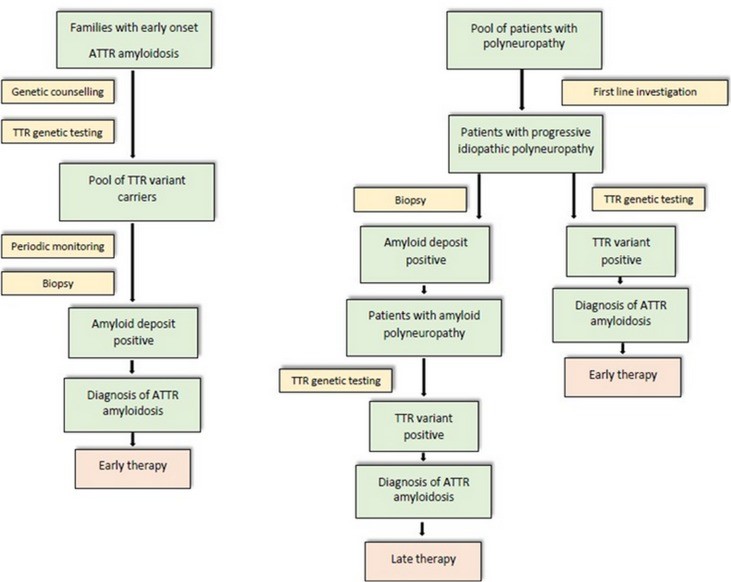
Table summarizing the varied clinical presentations and global distribution of common hATTR mutations, crucial for guiding diagnostic approaches based on genetic and geographical factors.
Clinical phenotype is strongly correlated with the specific TTR mutation. Certain variants, such as Val122Ile, Ile68Leu, Thr60Ala, and Leu111Met, predominantly manifest as cardiomyopathy, while others, like Ala97Ser and Ser50Arg, primarily present with a neurological phenotype. This genotype-phenotype correlation is vital for targeted hATTR diagnosis.
Age of onset also exhibits considerable variability and influences the phenotype and clinical course, along with geographic origin. Patients from endemic areas are typically described as having early-onset disease (age <50 years), whereas those from non-endemic regions often present with late-onset disease (age >50 years). These two groups, despite potentially sharing the same genotype, display differences in pathological and clinical features, further complicating hATTR diagnosis and requiring age-specific diagnostic considerations.
Early-onset patients in endemic regions usually develop a progressive sensory-motor and autonomic neuropathy, with an expected survival of approximately 10–20 years from onset. In contrast, late-onset Val30Met patients and non-Val30Met cases tend to have a more rapid and severe progression, often with significant cardiac involvement, and are associated with a median survival of approximately 7 years from disease onset. Prognosis is poorest for exclusively cardiac phenotypes (FAC), with a life expectancy of 2–5 years from symptom onset. These prognostic differences highlight the importance of early and accurate hATTR diagnosis for tailored management and patient counseling.
Symptoms of hATTR Amyloidosis and their Diagnostic Significance
Initial symptoms of hATTR amyloidosis, particularly in early-onset hATTR-Val30Met, can be systemic and non-specific, such as fatigue or unintentional weight loss. Recognizing these subtle early indicators is crucial for prompting further investigation and hATTR diagnosis.
The specific clinical picture of somatic neuropathy is influenced by the causative TTR mutation and age of onset. Early-onset hATTR-Val30Met typically presents with discomfort (numbness and neuropathic pain) in the feet, gradually extending proximally, accompanied by impaired pain and temperature sensation due to the involvement of unmyelinated and small myelinated fibers. Impairment of light touch, deep sensibility, and motor fibers usually occurs later in the disease course. In contrast, late-onset hATTR-Val30Met and other non-Val30Met variants exhibit more variable presentations, manifesting as sensory or sensorimotor symptoms starting distally in the lower or all four limbs, or even exclusively in the upper limbs, sometimes mimicking carpal tunnel syndrome. Differentiating these neuropathic patterns is a key step in hATTR diagnosis.
Autonomic impairment is frequently observed, especially in early-onset cases, and can manifest as gastrointestinal symptoms (constipation, diarrhea, alternating bowel habits, early satiety, nausea, vomiting), orthostatic hypotension, bladder dysfunction, and erectile dysfunction. Autonomic symptoms provide important diagnostic clues, particularly in combination with peripheral neuropathy.
Cardiac amyloidosis, often remaining latent for a prolonged period, eventually leads to progressive biventricular wall thickening, diastolic dysfunction due to loss of compliance, conduction disorders, and symptoms of congestive heart failure with preserved ejection fraction. Cardiac arrhythmias and/or conduction blocks are common in early-onset Val30Met or non-Val30Met variants (e.g., Glu89Gln). Cardiac evaluation is an integral part of hATTR diagnosis, especially given the often subtle initial cardiac involvement.
Renal involvement, typically presenting as nephrotic syndrome and/or progressive renal failure, occurs in approximately one-third of Portuguese Val30Met patients and in only 6% of sporadic hATTR cases. Ocular involvement is also frequent, with prevalence increasing with disease duration. Ocular disorders include dry eye syndrome, amyloid deposition on the iris or lens capsule, pupillary disorders, glaucoma, vitreous opacity, abnormal conjunctiva vessels, and amyloidotic retinal angiopathy. Systemic evaluation for renal and ocular manifestations is crucial for comprehensive hATTR diagnosis and staging.
Central nervous system (CNS) involvement, resulting from leptomeningeal amyloid angiopathy, has been described, sometimes in conjunction with eye impairment (oculoleptomeningeal amyloidosis). Leptomeningeal and meningovascular amyloidosis are rare forms caused by specific TTR variants, such as Ala25Thr or Tyr69His, but CNS impairment can also occur in advanced stages of hATTR-Val30Met, leading to stroke, subarachnoid hemorrhage, hydrocephalus, cerebellar ataxia, spastic paresis, seizures, hearing loss, and dementia. Although less common, CNS involvement represents a severe manifestation that must be considered in the differential hATTR diagnosis, particularly in advanced cases.
Diagnosis of hATTR Amyloidosis: Strategies for Timely and Accurate Identification
In endemic areas, where a positive family history is common, hATTR diagnosis is often straightforward and typically achieved within one year of symptom onset. In these regions, once a mutation is identified within a family, genetic counseling for asymptomatic carriers is essential. Regular follow-up of carriers is necessary to detect early disease signs, facilitating prompt initiation of anti-amyloid therapy.
hATTR diagnosis is significantly more challenging in non-endemic regions, where delays of 4–5 years are common due to factors such as negative family history and the heterogeneous clinical presentation mimicking other neuropathies.
Table 2. Frequent Misdiagnosis in Patients with Transthyretin Amyloidosis
| Phenotypes | Genotypes | Misdiagnosis |
|---|---|---|
| Length-dependent, small-fibre polyneuropathy and/or autonomic neuropathy | TTR Val30Met mutation (early-onset disease) | – Diabetic Polyneuropathy – Fibromyalgia – Immunoglobulin light-chain amyloidosis – Chronic digestive disease |
| All-fibre polyneuropathy | TTR Val30Met mutation (late-onset disease) and other TTR variants | – CIDP – Idiopathic axonal polyneuropathy – Lumbar spinal stenosis – Vasculitic peripheral neuropathy – Toxic peripheral neuropathy – Alcoholic neuropathy – Paraproteinemic neuropathy |
| Upper-limb-onset polyneuropathy | TTR Val30Met mutation (43%) and other TTR variants (Phe64Leu, Ser77Tyr, Tyr78Phe and Ile107Val) | – Carpal tunnel syndrome – Idiopathic polyneuropathy – CIDP – Paraneoplastic neuropathy – Cervical radiculopathy |
| Motor neuropathy | TTR Val30Met, Phe64Leu, Ile68Leu, Tyr78Phe, Val39Met and Ile107Val | – ALS – Motor CIDP – Motor neuropathy – Motor neuron disease |
| Cardiomyopathy | TTR variant (Thr60Ala, Leu111Met, Ile68Leu, Val122Ile) and wild-type ATTR | – Sarcomeric hypertrophic cardiomyopathies – Anderson-Fabry disease – Mitochondrial disorders (ie, Kearns–Sayre syndrome) – Danon disease – Noonan syndrome |
Table detailing common misdiagnoses for various hATTR amyloidosis phenotypes, emphasizing the need for careful differential diagnosis and heightened clinical awareness in practice.
Common misdiagnoses include idiopathic axonal polyneuropathy, chronic inflammatory demyelinating polyradiculoneuropathy (CIDP), lumbar spinal stenosis, diabetic neuropathies, Charcot-Marie-Tooth neuropathy, and motor neuron disease. In patients presenting with peripheral neuropathy of undetermined etiology, early consideration of associated clinical features, particularly cardiac involvement, is critical to suspect amyloidosis and pursue hATTR diagnosis.
Confirmation of hATTR diagnosis involves demonstrating amyloid deposits in a biopsy sample and/or detecting an amyloidogenic mutation through TTR gene sequencing.
Amyloid deposits can be identified in various tissues, including skin, nerve, myocardium, kidney, fat pad, labial salivary gland, or gastrointestinal mucosa. Tissue biopsy, ideally of an affected organ, is performed based on clinical suspicion. Congo red staining, which exhibits apple-green birefringence under polarized light, is used to reveal amyloid deposition. Once amyloid is confirmed, the precursor protein type must be identified, typically through immunohistochemistry or mass spectroscopy to confirm TTR as the precursor protein. However, the patchy distribution of amyloid fibrils can lead to false-negative or false-positive biopsy results.
Therefore, a negative biopsy does not exclude hATTR diagnosis. Biopsy sensitivity depends on the tissue sampled, the TTR mutation, and patient age. Multiple biopsies may be needed in some cases. In certain European countries, labial salivary gland biopsy is utilized to investigate amyloidosis in unexplained progressive axonal neuropathy. Nerve biopsy, while often a second-line investigation, has a sensitivity of approximately 80%. Amyloid visualization in muscle or abdominal fat biopsies shows variable sensitivity (14–83%).
Microscopic analysis of a sural nerve biopsy from a late-onset FAP patient, demonstrating Congo red staining for amyloid deposition (A) and ultrastructural confirmation of amyloid fibrils via electron microscopy (B), illustrating key pathological diagnostic techniques for hATTR.
Histopathological images of sural nerve biopsies from a late-onset FAP patient, revealing a significant reduction of myelinated fibers in semithin sections stained with toluidine blue (A) and complete loss of myelinated fibers in a subsequent biopsy four years later (B), while unmyelinated fibers are relatively preserved (C), highlighting nerve fiber degeneration in hATTR diagnosis.
Sural nerve biopsy analysis using various staining methods: Toluidine blue (A) and H&E (B) showing amyloid deposition, confirmed by Congo red (C), and immunofluorescence staining with anti-TTR (D), anti-kappa light chain (E), and anti-lambda light chain (F) to specifically identify TTR amyloid in hATTR diagnosis.
If clinical suspicion remains high despite a negative biopsy, TTR gene sequencing is recommended. Sanger sequencing is currently considered the gold standard for genetic confirmation of hATTR diagnosis.
Diagnostic algorithm flowchart for FAP in endemic and non-endemic areas, outlining a strategic approach to hATTR diagnosis incorporating clinical evaluation, biopsy, genetic testing, and ancillary investigations.
Once hATTR diagnosis is established, comprehensive investigations are necessary to assess the extent and severity of organ involvement.
Traditional nerve conduction studies are used to detect and quantify peripheral neuropathy. While skin biopsy remains the gold standard for diagnosing small fiber neuropathy, evaluating sudomotor function via electrochemical skin conductance, measuring heart rate variability, and testing for orthostatic hypotension are useful in assessing autonomic neuropathy.
Cardiac investigations in hATTR amyloidosis aim to detect infiltrative (hypertrophic) cardiomyopathy and conduction disorders that may necessitate prophylactic pacemaker implantation to mitigate sudden death risk. For mutations predominantly causing cardiac phenotypes, differential diagnosis with other cardiomyopathies is crucial. Useful cardiac assessments include echocardiogram, 24-hour Holter monitoring, echocardiography with strain imaging, cardiac magnetic resonance imaging (MRI), and intracardiac electrophysiological studies when available.
Scintigraphy using bone tracers such as 99mTc-DPD, 99mTc-HMDP, or 99mTc-PYP is a non-invasive method that can facilitate early hATTR diagnosis by identifying cardiac amyloid infiltration. It may serve as an alternative to cardiac biopsy in this context due to its high sensitivity and specificity, although potential low sensitivity for certain mutations like Phe64Leu should be considered.
Cardiac serum biomarkers, specifically brain natriuretic peptide (BNP) or its N-terminal prohormone (NT-proBNP) and cardiac troponins (T or I), are valuable for prognosis in amyloid cardiomyopathy. Elevated NT-proBNP levels, even in early stages, correlate with cardiac amyloid severity. High troponin levels are observed in severe or advanced disease stages.
Following genetic confirmation of hATTR diagnosis, ophthalmological assessment is essential. Frequency of evaluations depends on the severity of ocular manifestations and includes visual acuity and intraocular pressure measurement, Schirmer test, ocular fundus, and slit-lamp examination.
Renal evaluation, based on serum creatinine, proteinuria, microalbuminuria, and estimated glomerular filtration rate (eGFR), is also crucial. Serum cystatin C may offer a more sensitive marker for non-invasive GFR estimation compared to serum creatinine.
Finally, a modified body mass index (mBMI), adjusted for hypalbuminemia, serves as a nutritional status marker, reflecting the impact of gastrointestinal symptoms and malabsorption in hATTR patients. These comprehensive diagnostic evaluations are essential for effective patient management and prognostication following hATTR diagnosis.
Outcome Measures in hATTR-PN: Tools for Monitoring Disease Progression Post-Diagnosis
To assess disability extent in hATTR-PN patients post-hATTR diagnosis, clinical practice employs tools such as the FAP staging system and the Polyneuropathy Disability (PND) score. However, these scales provide a general disease status indicator and may not be sensitive enough to track short-term disease progression.
To better evaluate polyneuropathy aspects, clinical trials utilize Neuropathy Impairment Score (NIS)-based measures, enhancing the detection of treatment effects. The NIS score, a composite score assessing muscle weakness, sensory loss, and reflexes, and its subset, NIS-lower limbs (NIS-LL), have limitations, leading to modifications.
The NIS+7, combining clinical assessment with seven electrophysiological tests, was introduced in a 2013 trial to improve neuropathic impairment characterization and quantification. Recent trials by Alnylam and Ionis used modified NIS+7 (mNIS+7) variants – mNIS+7Alnylam and mNIS+7Ionis – specifically designed for hATTR amyloidosis clinical trials. These refined scores better quantify sensory abnormalities, nerve conduction abnormalities, and autonomic function, enhancing the precision of outcome monitoring after hATTR diagnosis.
Other valuable clinical scales include the Composite Autonomic Symptom Scale-31 (COMPASS-31) and the Compound Autonomic Dysfunction Test (CADT) questionnaires for autonomic symptom assessment; the Rasch-built Overall Disability Scale (R-ODS) for activities of daily living evaluation; the Norfolk Quality of Life-Diabetic Neuropathy (QoL-DN) questionnaire for quality of life estimation; and the 6-minute walk test, 10-meter walk test, and handgrip strength test for motor function assessment. Instrumental examinations like Sudoscan also prove useful in monitoring disease progression in late-onset hATTR. Integrating these outcome measures into clinical follow-up post-hATTR diagnosis is crucial for comprehensive patient management.
Considering the array of available scales and questionnaires, practical application of all in clinical practice is challenging. Clinicians might prioritize FAP staging system, PND, and NIS-LL scores for daily clinical use, while reserving NIS+7 and its variants for research protocols. For autonomic involvement, CADT questionnaire and Sudoscan may represent optimal tools for assessment and monitoring following hATTR diagnosis.
Therapy for hATTR Amyloidosis: Management Strategies Post-Diagnosis
Management of hATTR amyloidosis, a systemic disease, requires a multidisciplinary approach following hATTR diagnosis. Care encompasses anti-amyloid therapy to inhibit further amyloid production/deposition, symptomatic therapy, and treatment for cardiac, renal, and ocular involvement, including potential heart or kidney transplantation in advanced disease.
Initiating timely treatment post-hATTR diagnosis is crucial to delay disease progression. Systematic monitoring of asymptomatic TTR carriers is essential for early disease detection and prompt anti-amyloid therapy initiation. Recent years have witnessed significant therapeutic advancements for hATTR amyloidosis, particularly for early-stage disease, although effective treatments for advanced stage 3 hATTR-PN remain limited.
Overview of therapeutic strategies for hATTR with polyneuropathy, illustrating the range of treatment options available post-hATTR diagnosis, from liver transplantation to gene-silencing therapies.
Liver Transplantation in hATTR Management
Liver transplantation (LT), introduced in 1990, was the first therapeutic approach for hATTR amyloidosis, aiming to prevent amyloid formation by replacing the primary TTR production site. LT outcomes are influenced by patient characteristics like age, disease severity at surgery, and TTR mutation. A retrospective analysis of 1940 patients from the FAP World Transplant Registry showed a 20-year survival rate of 55.3%, with cardiovascular complications as the leading cause of death (22%). While LT replaces mutant TTR with wild-type protein, it may not halt progressive cardiac amyloid deposition due to pre-existing fibrils potentially templating wild-type TTR polymerization, contributing to cardiomyopathy. Despite its limitations, LT remains a consideration in select hATTR diagnosis cases, particularly in early stages.
TTR Stabilizers in hATTR Therapy
TTR tetramer stabilizers are designed to stabilize the normal circulating TTR form, preventing dissociation and conformational changes leading to amyloid aggregation. However, they do not inhibit ATTR “seeding,” potentially explaining clinical trial results. Two TTR stabilizers have been evaluated in controlled hATTR amyloidosis trials:
Tafamidis for hATTR-PN and hATTR-CM
Tafamidis, an oral kinetic TTR stabilizer, is approved in Europe and selected countries for stage 1 symptomatic hATTR-PN in adults. A phase III trial involving 128 patients, mainly Portuguese with early-onset ATTR-Val30Met and mild neuropathy, showed tafamidis stabilized TTR in 98% of patients and halted neuropathy progression in 60% (vs. 38% placebo), maintaining quality of life. Tafamidis’s efficacy and safety in late-onset hATTR have been assessed in open-label studies, showing tolerability but no prevention of disability progression at 1 year. More recently, based on the ATTRACT trial, tafamidis was FDA-approved for hATTR-CM treatment, demonstrating reduced mortality and cardiovascular hospitalizations in symptomatic hATTR-CM and ATTRwt amyloid cardiomyopathy patients. Tafamidis represents a significant pharmacological option post-hATTR diagnosis.
Diflunisal as a TTR Stabilizer
Diflunisal, a nonsteroidal anti-inflammatory drug (NSAID), also acts as a TTR stabilizer. A phase III study in late-onset hATTR amyloidosis patients with various TTR mutations and high NIS at baseline showed that 2-year diflunisal treatment significantly reduced neuropathy progression and preserved quality of life compared to placebo. However, treatment discontinuation due to disease progression and liver transplantation was common, and long-term NSAID use carries risks of gastrointestinal, renal, and cardiac adverse events. Diflunisal is another therapeutic consideration following hATTR diagnosis, albeit with caution regarding long-term safety.
Gene-Silencing Therapies in hATTR Management
Novel gene-silencing therapies, patisiran (Onpattro) and inotersen (Tegsedi), are approved in the US and Europe for stage 1 or 2 polyneuropathy in adults with hATTR amyloidosis. These drugs reduce both variant and wild-type TTR hepatic production by targeting mRNA. Long-term studies are needed to assess gene-silencing effects on TTR’s functional roles, particularly vitamin A transport. These therapies represent a paradigm shift in hATTR diagnosis and management.
RNA Interference with Patisiran and Revusiran
RNA interference (RNAi) employs small interfering RNAs (siRNAs) to inhibit specific gene expression by degrading targeted mRNAs. siRNAs are encapsulated in lipid nanoparticles for liver delivery after intravenous administration. RNAi therapy for hATTR amyloidosis has shown promise in preclinical and clinical trials. Preclinical models showed significant TTR deposit reduction in peripheral tissues with RNAi-mediated TTR suppression. A phase I study of patisiran demonstrated dose-dependent TTR reduction in healthy volunteers and hATTR patients. A phase II study in stage 1 or 2 hATTR-PN patients showed up to 87% mean TTR level reduction with patisiran treatment. An open-label extension study showed neuropathy impairment stability over 24 months. The phase III APOLLO study confirmed patisiran efficacy and safety over 18 months, with mNIS+7Alnylam score improvement and benefits in quality of life, motor strength, disability, gait speed, nutritional status, and autonomic symptoms. Common adverse events were mild to moderate peripheral edema and infusion-related reactions. Revusiran, another RNAi drug for hATTR amyloidosis, was discontinued in phase III trials due to increased mortality in the treatment group. Patisiran remains a significant advancement in hATTR diagnosis and treatment.
Antisense Oligonucleotides with Inotersen
Inotersen (IONIS-TTRRx) is a second-generation antisense oligonucleotide (ASO) inhibiting hepatic production of both mutant and wild-type TTR by targeting and degrading TTR mRNA. Robust TTR level reduction was observed in animal models and healthy humans treated with inotersen. The phase III NEURO-TTR trial assessed inotersen efficacy and safety over 15 months in stage 1 or 2 hATTR-PN patients, showing significant improvement in mNIS+7Ionis score and Norfolk QoL-DN questionnaire scores compared to placebo, irrespective of TTR mutation or disease stage. 36.6% of inotersen-treated patients remained stable or improved on mNIS+7 score at 15 months versus 19.2% in the placebo arm. Serious adverse events included glomerulonephritis and thrombocytopenia, with one death associated with thrombocytopenia, leading to enhanced monitoring for these conditions. Inotersen is FDA and EMA approved for stage 1 or 2 hATTR-PN treatment, offering another vital therapeutic option post-hATTR diagnosis.
Emerging Therapeutic Approaches for hATTR
Emerging therapeutic approaches include agents that stabilize TTR, such as epigallocatechin-3-gallate (EGCG) from green tea, which may enhance TTR tetramer stabilization and reduce TTR deposition. Monoclonal antibodies targeting non-native TTR forms are another potential therapy, selectively binding misfolded TTR and inhibiting fibril formation in vitro. A phase I–II study is ongoing to evaluate AKCEA-TTR-LRx (ION-682884), an antisense inhibitor of transthyretin production. A phase III study is recruiting patients to assess vutrisiran (ALN-TTRSC02), an RNAi drug administered subcutaneously every 3 months, comparing it to patisiran. These emerging therapies hold promise for future advancements in hATTR diagnosis and treatment.
Conclusion: Advancing hATTR Diagnosis and Management for Improved Patient Outcomes
hATTR amyloidosis is a severe, genetically and clinically diverse multisystem disease with global distribution. Recent clinical trial results have broadened therapeutic options to include TTR stabilizers and gene silencers, reducing liver transplantation utilization. Gene-silencing therapies are of increasing interest, potentially altering the disease’s natural history across stages, although long-term effects remain under investigation, particularly concerning persistent transthyretin production in the brain and eye. Future challenges include finding effective therapies for advanced disease, assessing amyloid deposit clearance with monoclonal antibodies, exploring curative gene replacement therapy, and evaluating combined therapies and disease-modifying therapies for pre-symptomatic individuals. Continued advancements in hATTR diagnosis and therapeutic strategies are crucial for improving patient outcomes and quality of life.
Abbreviations
9mTc-DPD, technetium-99m radiolabeled 2,3-dicarboxypropane-1,1-diphosphonate; 99mTc-HMDP, technetium-99m radiolabeled hydroxymethylene diphosphonate; 99mTc-PYP, technetium-99m radiolabeled pyrophosphate; ASO, antisense oligonucleotide; ATTRwt, wild-type transthyretin amyloidosis; BNP, brain natriuretic peptide; CADT, Compound Autonomic Dysfunction Test; CNS, central nervous system; COMPASS-31, Composite Autonomic Symptom Scale-31; CSF, cerebrospinal fluid; EGCG, epigallocatechin-3-gallate; eGFR, estimated glomerular filtration rate; EMA, European Medicines Agency; FAC, familial amyloid cardiomyopathy; FAP, familial amyloid polyneuropathy; FDA, Food and Drug Administration; GalNAc, N-acetylgalactosamine; hATTR, hereditary transthyretin amyloidosis; hATTR-CM, hereditary transthyretin amyloidosis with cardiomyopathy; hATTR-PN, hereditary transthyretin amyloidosis with polyneuropathy; LT, liver transplantation; mBMI, modified body mass index; mNIS+7, modified Neuropathy Impairment Score +7; MRI, magnetic resonance imaging; NIS, Neuropathy Impairment Score; NIS+7, Neuropathy Impairment Score +7; NIS-LL, Neuropathy Impairment Score-Lower Limbs; Norfolk QoL-DN, Norfolk Quality of Life-Diabetic Neuropathy; NSAID, nonsteroidal anti-inflammatory drug; NT-proBNP, N-terminal prohormone of brain natriuretic peptide; PND, Polyneuropathy Disability score; QoL, quality of life; RNAi, RNA interference; RNase H1, ribonuclease H1; R-ODS, Rasch-built Overall Disability Scale; siRNAs, small interfering RNAs; T4, thyroxine; TTR, transthyretin.
Disclosure
Dr Luigetti received financial grants (honoraria and speaking) from Akcea, Alnylam and Pfizer, and travel grants from Pfizer, Kedrion and Grifols; Dr Bisogni received financial grants (honoraria and speaking) from Alnylam, and travel grants from Pfizer, and Grifols; Dr Romano received travel grants from Akcea and Pfizer; Dr Di Paolantonio received travel grants from Akcea and Pfizer; Dr Sabatelli received financial grants (honoraria and speaking) from Akcea. The authors report no other conflicts of interest in this work.