Abstract
This clinical review offers a practical diagnostic approach to hemochromatosis from two experienced clinicians. Advancements in genetic testing and extensive population studies have significantly improved our understanding of the disease’s clinical presentation and the diagnostic value of biochemical iron tests for disease detection and severity assessment. Liver biopsy is now more valuable for prognosis than initial diagnosis. We present a cost-effective, stepwise diagnostic algorithm grounded in current evidence, emphasizing early diagnosis to facilitate phlebotomy therapy and prevent cirrhosis.
Keywords: Hemochromatosis, HFE, Iron Overload, Diagnosis Criteria
Introduction to Hemochromatosis Diagnosis
Hereditary hemochromatosis (HH) is frequently cited as an ideal candidate for widespread screening due to readily available diagnostic tools and effective treatments, coupled with a significant genetic prevalence, particularly in Caucasian populations (approximately 1 in 227) [1]. However, large-scale population screening initiatives have revealed a complex landscape, identifying numerous asymptomatic individuals and control groups without HFE mutations exhibiting similar symptoms [1–3]. The natural progression of untreated hemochromatosis remains incompletely understood and is unlikely to be fully elucidated through clinical trials. This review aims to summarize current perspectives on hemochromatosis screening and diagnosis, providing a clear pathway for clinicians.
Defining Iron Overload in Hemochromatosis
Identifying iron overload conditions is a critical public health concern, as the organ damage induced by excessive iron accumulation is often preventable with timely diagnosis and intervention. While iron overload can stem from various causes, hereditary hemochromatosis represents the majority of cases in North America. HH is an autosomal recessive genetic disorder resulting from mutations in the HFE gene, located on chromosome 6 [4]. The most prevalent mutation, C282Y, arises from a G to A missense substitution, replacing cysteine with tyrosine at position 282 of the HFE protein. This C282Y mutation is a defining genetic marker of HH.
In its fully expressed form, homozygous C282Y mutation leads to progressive iron accumulation in vital organs, notably the liver, pancreas, heart, joints, and endocrine glands, ultimately causing structural and functional damage [5]. Hemochromatosis is unique among genetic conditions because the life-threatening consequences of iron overload, such as liver damage (fibrosis or cirrhosis), diabetes mellitus, and cardiomyopathy, often manifest later in life, typically in middle age.
This extended latency period between genetic predisposition and the onset of severe symptoms presents a crucial window for early diagnosis and treatment. The challenge of early diagnosis is amplified by the high prevalence of the homozygous C282Y mutation in North American Caucasians (1 in 227) [1 and the accessibility of phenotypic and genotypic markers for primary care physicians. Despite these diagnostic advancements, ongoing debates surround the natural history of HH, questioning the broad application of these techniques in generally healthy populations. Unselected population studies have challenged the previously held belief of 100% penetrance for the homozygous mutation [[1](#b1-cjg200535],[2](#b2-cjg200535],6. It is now understood that up to 50% of C282Y homozygotes may not develop clinically significant iron overload even by middle age. This realization prompts a re-evaluation of the cost-effectiveness of screening strategies utilizing both phenotypic iron markers and gene mutation analysis [[7](#b7-cjg200535],8. However, the benefits of family screening and iron overload assessment in patients with liver disease, diabetes mellitus, unexplained cardiac issues, arthropathies, and male sexual dysfunction remain strongly supported [9].
Classifying Iron Overload Conditions in Differential Diagnosis
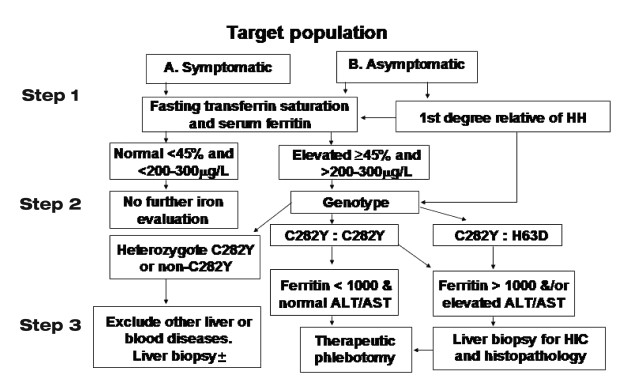
While this discussion encompasses the broader evaluation of all iron overload conditions, the primary focus remains on hereditary hemochromatosis. The majority of HH cases are HFE-related. However, emerging research has identified familial HH cases exhibiting classic phenotypic criteria but lacking detectable HFE gene mutations [[10](#b10-cjg200535],11. Accumulating case studies are revealing alternative gene mutations that may account for a portion of the estimated 10% of non-HFE mutations in families with typical HH. The diagnostic algorithm for HH, endorsed by leading gastroenterology societies, provides a structured approach for differentiating HFE-related HH, non-HFE-related HH, and secondary iron overload forms (Figure 1) [12.
Figure 1). Diagnostic Algorithm for Hemochromatosis Investigation.
 Diagnostic algorithm for hemochromatosis investigation, including blood studies, mutation analysis and liver biopsy steps
Diagnostic algorithm for hemochromatosis investigation, including blood studies, mutation analysis and liver biopsy stepsClinical Presentation: Recognizing Hemochromatosis Features
The classic clinical features of HH, initially described in the late 19th century and comprehensively detailed by Sheldon in 1935 [13], represent the advanced syndrome with significant end-organ damage. This includes cirrhosis, diabetes mellitus, skin pigmentation, cardiomyopathy, hypogonadism, and arthropathy. Early studies focusing on symptomatic probands and their families indicated that most cases presented with these advanced complications, often with established cirrhosis and diabetes [[14](#b14-cjg200535],15].
Increased awareness and the identification of asymptomatic individuals with HH have shifted the clinical landscape. A larger proportion of diagnosed patients are now younger, without cirrhosis or diabetes, and exhibit a significantly improved prognosis [15–17]. Longitudinal studies spanning five decades have demonstrated that diagnosing and treating HH before these complications develop is associated with a normal life expectancy [15].
Iron overload complications contribute to at least 60% of HH-related deaths, with liver disease complications (liver failure from cirrhosis or hepatocellular carcinoma) accounting for three-quarters of these deaths [[15](#b15-cjg200535],18]. Cirrhosis, once established, is rarely reversible, and hepatocellular carcinoma remains a risk in cirrhotic HH patients, even after successful iron removal via phlebotomy. Diabetes mellitus is the second leading cause of death in HH [15]. While some evidence suggests improved diabetes management in insulin-dependent patients with phlebotomy, data supporting reduced diabetes-related mortality after disease onset are lacking. Similarly, the benefit of iron reduction therapy in preventing cardiomyopathy in HH is presumed but not definitively proven. Furthermore, evidence is lacking to support early diagnosis and treatment in preventing less severe complications like arthropathy and hypogonadism. Arthritis, specifically, typically does not improve with iron depletion therapy.
Large pedigree studies over 40-50 years indicated an approximate 70% prevalence of cirrhosis and/or diabetes in HH [[15](#b15-cjg200535],16. However, these complication rates are significantly lower in individuals diagnosed through pedigree analysis or in recent decades. Population screening studies report cirrhosis prevalence as low as 1% to 5% [[19](#b19-cjg200535],20, and diabetes prevalence is not consistently identified as uniquely associated with hemochromatosis compared to control populations [[1](#b1-cjg200535],2. Studies based on mutation analysis of unselected populations show that only a minority of C282Y homozygotes are symptomatic, and very few exhibit chronic liver disease or diabetes unless additional risk factors like alcohol abuse, hepatitis C, or obesity are present [[21](#b21-cjg200535],22. This “changing scene” [16] may reflect variations in iron accumulation rates among homozygotes or previously unrecognized differences in susceptibility to iron toxicity across families. Historically, HH progression was categorized into stages based on age and iron storage levels [[15](#b15-cjg200535],[23](#b23-cjg200535],24. Women typically manifest symptoms 5-10 years later than men, likely due to iron loss during reproductive years. The concept of the hepatic iron index (HII) and critical hepatic iron levels linked to fibrosis and cirrhosis emerged from this understanding of age-related iron accumulation [[25](#b25-cjg200535],26. It’s now recognized that a small subset of homozygotes do not exhibit the previously considered diagnostic HII (greater than 1.9 μmol/g/year), and other iron overload types can present with HII exceeding this value [23.
Step-by-Step Hemochromatosis Diagnostic Algorithm
Hemochromatosis diagnosis is considered in various clinical scenarios: symptomatic patients, asymptomatic first-degree relatives of diagnosed individuals, asymptomatic patients with incidental findings like elevated liver enzymes or iron markers, or those being evaluated for associated conditions such as diabetes, arthropathy, or cardiomyopathy, and in the general population. The generally accepted minimum diagnostic criteria for HH are increased iron stores and/or C282Y/C282Y or C282Y/H63D HFE gene mutations. While the diagnostic significance of gene mutation analysis alone is debated, the necessity for treatment is ultimately determined by assessing the extent of iron loading.
Diagnostic accuracy assessments for phenotypic screening methods are derived from both unselected populations [27] and families of C282Y homozygotes [28]. Family-based studies may have diagnostic incorporation bias due to elevated iron tests being part of the inclusion criteria [29]. Based on available data and cost-effectiveness analyses, gastroenterological societies have developed a diagnostic algorithm for evaluating suspected HH cases [12].
Step 1: Initial Blood Iron Studies for Hemochromatosis
Given the consensus that hemochromatosis diagnosis primarily relies on phenotypic evidence of iron overload, with genotypic analysis as secondary confirmation, the initial diagnostic approach involves indirect markers of iron stores: transferrin saturation (TS), unsaturated iron binding capacity (UIBC), and serum ferritin. TS is calculated as the ratio of serum iron to total iron binding capacity, often derived from the sum of serum iron and UIBC or from transferrin concentration. Ideally, TS should be measured after overnight fasting to minimize circadian and postprandial variations. Nonfasting samples increase false-positive rates [30]. Sensitivity, specificity, and positive predictive value of TS vary depending on the population studied. A TS cutoff of 45% or higher is often used to optimize predictive accuracy for iron overload detection [[1](#b1-cjg200535],31]. However, population studies have reported lower sensitivity for TS greater than 50% [2, likely due to the prevalence of non-expressing C282Y homozygotes [32]. UIBC has demonstrated similar or slightly superior performance to TS in detecting C282Y homozygotes at a lower cost. A UIBC below 26 μmol/L exhibited 90% sensitivity, 90% specificity, and a 2.2% positive predictive value for detecting C282Y homozygotes [27]. Men generally show greater biochemical iron abnormalities and clinical symptom expression in hemochromatosis.
Studies of HH families have shown serum ferritin concentration to correlate well with body iron stores [[28](#b28-cjg200535],33 (Table 1). Given the progressive iron accumulation in most HH patients, some young homozygotes may present with normal serum ferritin but elevated TS. However, serum ferritin generally provides additional confirmation of elevated TS significance in homozygotes. A normal serum ferritin combined with TS below 45% has a 97% negative predictive value for excluding iron overload [28. A large study in a primary care population (41,038 individuals screened) reported serum ferritin above 250 μg/L in males and 200 μg/L in females to be positive in 76% and 54% of C282Y homozygotes, respectively [2].
TABLE 1. Prevalence of Elevated Ferritin in C282Y Homozygotes from Screening Studies
| Population sample (reference) | Number screened | C282Y homozygotes | Ferritin Prevalence (Male, >300 μg/L or 250 μg/L*) | Ferritin Prevalence (Female, >200 μg/L*) |
|---|---|---|---|---|
| Electoral roll [56] | 1064 | 1 in 213 | 1/1 (100%) | 2/4 (50%) |
| Ambulatory care [57] | 1653 | 1 in 276 | 1/1 (100%) | 2/5 (40%) |
| Epidemiology survey [6] | 3011 | 1 in 188 | 5/5 (100%) | 3/7 (43%) |
| Ambulatory care [2] | 41,038 | 1 in 271 | 55/72 (76%)* | 43/79 (54%) |
| Ambulatory care [19] | 65,238 | 1 in 220 | 90%† | 60%* |
| Ambulatory care (HEIRS) [1] | 101,168 | 1 in 227 in Caucasians | 88% | 57% |
[*Ferritin >250 μg/L was the lower limit for males in reference 2; †In this large study, genetic testing was only done in cases with an elevated transferrin saturation. The lower expression in women was estimated from observed versus expected prevalence of iron overload. HEIRS Hemochromatosis and Iron Overload Screening study]
Nonspecific elevations in serum ferritin can occur in various conditions unrelated to iron overload. Therefore, isolated elevated ferritin without elevated TS may be nonspecific. However, iron overload can be present with elevated ferritin and normal TS, particularly in non-HFE-related iron overload [10]. Significant ferritin elevations (greater than 1000 μg/L) with normal TS and no apparent cause may warrant liver biopsy to assess for iron overload. Furthermore, serum ferritin serves as a predictor of fibrosis or cirrhosis in confirmed HH. Studies indicate that serum ferritin below 1000 μg/L accurately predicts the absence of cirrhosis, regardless of disease duration [34–36].
Step 2: HFE Mutation Analysis for Hemochromatosis Genotyping
Since phenotypic expression is paramount for determining the need for iron overload treatment, individuals with fasting TS below 45% and normal serum ferritin require no further hemochromatosis evaluation. Elevated iron markers prompt genotypic testing, specifically for HFE gene mutations C282Y and H63D, which account for over 90% of phenotypic HH cases. These figures were summarized at a National Institutes of Health conference on HH [37]. C282Y homozygotes or C282Y:H63D compound heterozygotes with elevated iron markers but without additional liver disease risk factors (viral hepatitis, alcohol abuse) or signs of significant hepatic damage may be offered therapeutic phlebotomy without further evaluation. Studies suggest that individuals under 40 with no hepatomegaly, normal alanine aminotransferase, no history of viral hepatitis or alcohol abuse, and serum ferritin below 1000 μg/L have minimal risk of liver fibrosis or cirrhosis [34–36].
Rare cases of iron overload are associated with mutations in other iron-related genes like ferroportin, hemojuvelin, hepcidin, ceruloplasmin, and transferrin receptor 2 [[10](#b10-cjg200535],[11](#b11-cjg200535],38]. Population prevalence of these mutations is not well-defined, and genetic testing is more complex due to diverse mutations across large gene regions. Commercial testing for these rare conditions is less likely due to their complexity and low prevalence.
For first-degree relatives of known HH probands, mutation analysis is recommended for those over 20 years to enable regular monitoring of C282Y homozygotes or C282Y:H63D compound heterozygotes using phenotypic markers. Cost-effectiveness analysis suggests that assessing genetic risk in children of probands is best achieved by spouse mutation analysis [39, although nonpaternity is a potential consideration.
Step 3: Liver Biopsy and Phlebotomy Therapy in Hemochromatosis Management
Based on current evidence, liver biopsy is indicated in specific hemochromatosis scenarios:
- C282Y homozygotes or C282Y:H63D compound heterozygotes with hepatomegaly, abnormal liver tests, liver disease risk factors, or serum ferritin above 1000 μg/L.
- C282Y heterozygotes with indirect iron markers suggesting extreme iron overload (e.g., serum ferritin > 1000 μg/L).
- Suspected non-HFE-related iron overload based on elevated serum ferritin (> 1000 μg/L).
In these situations, liver biopsy confirms cirrhosis presence, which carries morbidity and mortality risks requiring surveillance and management [15]. It also provides tissue for histological evaluation and biochemical iron store measurement. Earlier studies suggested a cirrhosis risk when hepatic iron concentration exceeded 300 μmol/g dry weight [20–22]. However, it’s now recognized that fibrosis can develop earlier in homozygotes with other risk factors, and some individuals exceed this threshold without developing cirrhosis [40. The HII is also considered a less stringent diagnostic criterion. Cirrhosis confirmation guides ongoing monitoring for complications.
If liver biopsy is deemed unnecessary based on the absence of these criteria, therapeutic phlebotomy serves as the ultimate assessment of total body iron stores. Symptomatic HH patients typically require 5-20g of iron removal for depletion, while presymptomatic cases require less (3-5g). In summary, evidence supports liver biopsy as a valuable prognostic tool in HH, guiding clinical care when cirrhosis is detected. However, recognizing potential patient reluctance, it remains an optional procedure to be discussed. Suspected cirrhosis warrants follow-up guidelines, including endoscopy for esophageal varices and abdominal ultrasound with alpha-fetoprotein for hepatocellular carcinoma screening.
General Population Screening for Hemochromatosis: Considerations and Controversies
Deciding on general population screening for HH requires careful evaluation of phenotypic-genotypic correlations, particularly how HFE mutation carriers manifest abnormal iron markers and develop clinical iron overload. Genetic testing advances have identified typical C282Y homozygotes without iron overload, especially in population screening and pedigree investigations. C282Y homozygotes can be categorized into three groups:
1. C282Y Homozygotes with Normal Iron Markers
The long-term risk of iron overload in this group is uncertain. Most cases are identified in adults, and progression to abnormal iron tests is rare. Following up this group, potentially representing up to 50% of C282Y homozygotes [[32](#b32-cjg200535],41, poses a significant cost, especially if they never develop iron overload. Long-term follow-up studies have shown that not all C282Y homozygotes develop progressive iron overload [[42](#b42-cjg200535],43. The Copenhagen Heart Study, monitoring C282Y homozygotes for up to 25 years, observed only slight increases in iron markers without overt disease [43. However, hepatic fibrosis can remain silent until cirrhosis complications arise.
2. C282Y Homozygotes with Elevated TS and/or Ferritin
This group often presents for other medical reasons, frequently asymptomatic or with nonspecific symptoms like fatigue, where attributing symptoms to hemochromatosis is uncertain. Elevated TS may be due to low transferrin in other chronic conditions. The natural history of mild, untreated cases is not well-established, and historical data include cases with non-progressive iron marker elevation [44. Most newly diagnosed cases fall into this category and should avoid organ damage with appropriate treatment.
3. C282Y Homozygotes with Significant Iron Overload and Organ Damage
These patients were dominant in tertiary referral center studies, potentially overestimating hemochromatosis morbidity. Serum ferritin correlates strongly with liver cirrhosis. Cirrhosis risk in C282Y homozygotes can be predicted by a clinical index using serum ferritin > 1000 μg/L, platelet count < 200×109/L, and AST > 40 U/L [35]. Diabetes is common in cirrhotic patients.
A key question is whether population screening detects clinically significant disease. A large Norwegian screening study (65,238 participants) with liver biopsies in 147 cases found cirrhosis in only four men and no women [19. The HEIRS study reported a minimum 0.66% liver fibrosis risk in C282Y homozygotes [45. An Australian study (3011 participants) with six liver biopsies identified one cirrhosis and two fibrosis cases [6. A San Diego screening study (41,038 participants) suggested a low (around 1%) prevalence of life-threatening hemochromatosis complications in C282Y homozygotes [2. This study also highlighted the difficulty of symptom attribution in hemochromatosis due to common symptoms like fatigue and arthralgias in the general population. The discrepancy between morbidity in referred vs. screened patients is not unique to hemochromatosis. Neonatal screening studies in France used reverse cascade screening to detect C282Y homozygotes in newborn families [46, raising ethical questions about genetic testing for late-onset diseases.
Mild to moderate iron overload can occur in compound heterozygotes (C282Y/H63D) [47 or H63D homozygotes [48, but organ damage is rare without other risk factors. Most individuals with these genotypes have normal iron studies [1, and iron overload can occur in individuals with normal genotypes due to other causes.
Target Populations for Hemochromatosis Screening
Large studies have refined target populations for iron overload screening. Currently, men of Northern European ancestry are considered the highest risk group for HFE-related hemochromatosis with iron overload. The HEIRS study (101,168 participants) demonstrated low HFE mutation prevalence in other ethnicities [27, but also showed higher TS and ferritin elevations in North Americans of Asian descent [1. African-American iron overload has been reported, but population studies have not yet identified patients or pedigrees [49. Economic models suggest hemochromatosis screening could be cost-effective even if only 20% of patients develop life-threatening complications [7, although this may be an overestimate given poorly defined natural history in unscreened, untreated groups. Concerns about genetic discrimination have been raised, but studies have not shown adverse psychosocial effects or discrimination [50–54. A large Australian screening study demonstrated high acceptance of genetic testing as the initial screening tool [55. Widespread population screening for hemochromatosis is unlikely to be recommended in North America [29, but targeted screening in high-risk populations warrants further investigation.
Summary: Key Hemochromatosis Diagnosis Criteria
This review aims to provide evidence-based data supporting early hemochromatosis detection to prevent organ damage and increased mortality. Further management aspects, including treatment and complication monitoring, are beyond this paper’s scope. Typically, patients are referred to hematologists or hepatologists for specialized care at that stage.
Acknowledgements
We dedicate this manuscript to the memory of Dr. David Brandhagen (Mayo Clinic, Rochester, Minnesota, USA), with whom we shared many insightful discussions on hemochromatosis.
References
1 цит. по: Beutler E, Felitti VJ, Koziol JA, et al. Penetrance of hemochromatosis. N Engl J Med 2002;346:354-358.
2 цит. по: Allen KJ, سؤالي J, Gurrin LC, et al. HFE C282Y hemochromatosis and iron overload phenotype in 41,038 people of northern European ancestry. Blood 2008;111:3071-3077.
3 цит. по: McCune CA, Raval M, Yu C, et al. Population prevalence of HFE mutations. Am J Hematol 2007;82:483-485.
4 цит. по: Feder JN, Gnirke A, Thomas W, et al. A novel MHC class I-like gene is mutated in patients with hereditary haemochromatosis. Nat Genet 1996;13:399-408.
5 цит. по: Edwards CQ, Kushner JP. Screening for hemochromatosis. Semin Liver Dis 2003;23:221-232.
6 цит. по: Crawford DH, Stuart K, ব্যাপারী M, et al. Population-based screening for HFE hemochromatosis: Prevalence and morbidity in an Australian community. Med J Aust 2004;180:617-621.
7 цит. по:꽈ری A, Harris E, Fano V, et al. Cost-effectiveness of genetic screening for hemochromatosis. Am J Med Genet 2000;96:713-722.
8 цит. по: Skjaerpe PA, Berg JP, Tollersrud O, et al. Cost-effectiveness of screening for hemochromatosis with transferrin saturation. Scand J Clin Lab Invest 2004;64:115-124.
9 цит. по: Bacon BR, Adams PC, Burke W, et al. Hereditary hemochromatosis. Gastroenterology 2011;140:1728-1733.
10 цит. по: Pietrangelo A. Hereditary hemochromatosis – a new look at an old disease. N Engl J Med 2004;350:2383-2397.
11 цит. по: Beutler E, Barton JC. HFE hemochromatosis. Annu Rev Med 2006;57:25-38.
12 цит. по: Bacon BR, Tavill AS, Bonkovsky HL, et al. Diagnosis and management of hemochromatosis: 2011 Practice Guideline by the American Association for the Study of Liver Diseases. Hepatology 2011;54:328-343.
13 цит. по: Sheldon JH. Haemochromatosis. London: Oxford University Press, 1935.
14 цит. по: Bomford A, Williams R. Long term results of venesection therapy in haemochromatosis. Q J Med 1976;45:611-623.
15 цит. по: Niederau C, Fischer R, Sonnenberg E, et al. Survival and causes of death in cirrhotic and in noncirrhotic patients with primary hemochromatosis. N Engl J Med 1985;313:1256-1262.
16 цит. по: Powell LW, Dixon JL, Hewett DG. Clinical aspects of hemochromatosis. Gut 2004;53:531-540.
17 цит. по: Adams PC, Valberg LS. Evolving expression of hemochromatosis in homozygous siblings. Hepatology 1996;23:1198-1202.
18 цит. по: Bradbear RA, Bain C, Siskind V, et al. Cohort study of internal malignancy in genetic hemochromatosis and other chronic nonalcoholic liver diseases. J Natl Cancer Inst 1985;75:81-84.
19 цит. по: Asberg A, Thorstensen K, Marthen E, et al. Body iron stores in HFE hemochromatosis: Assessment by serum ferritin and estimated hepatic iron stores in a population-based study of 65,238 persons. Gut 2001;49:717-723.
20 цит. по: Bassett ML, Halliday JW, Powell LW. Value of hepatic iron measurement in early hemochromatosis and determination of the critical iron level associated with fibrosis. Hepatology 1986;6:24-29.
21 цит. по: Moirand R, Jouanolle AM, Brissot P, et al. Liver iron concentration in HFE hemochromatosis: A re-evaluation of the diagnostic threshold and the relationship to cirrhosis. Gastroenterology 1997;112:555-559.
22 цит. по: Summers KM, Halliday JW, Powell LW. Identification of homozygous hemochromatosis subjects by measurement of serum ferritin in a large population screen. Clin Chim Acta 1983;130:17-25.
23 цит. по: Deugnier YM, Turlin P, Powell LW, et al. Different mechanisms of hepatic iron overload in genetic hemochromatosis and alcoholic liver disease. Hepatology 1993;17:27-32.
24 цит. по: Valberg LS, Lloyd DA, Whitman W, et al. Clinical and biochemical expression of the genetic defect in hemochromatosis. Gastroenterology 1985;89:1305-1313.
25 цит. по: Bassett ML, Halliday JW, Ferris RA, et al. Diagnosis of hemochromatosis in young people: Predictive accuracy of biochemical screening tests. Gastroenterology 1984;87:628-633.
26 цит. по: Summers KM, Powell LW, Halliday JW. Hepatic iron overload in alcoholic liver disease. Hepatology 1983;3:68-75.
27 цит. по: Adams PC, Reboussin DM, Barton JC, et al. Evaluation of serum transferrin saturation and unsaturated iron-binding capacity in genetic hemochromatosis probands and population controls. Am J Gastroenterol 2000;95:2479-2485.
28 цит. по: Edwards CQ, Griffen LM, Goldgar DE, et al. Phenotypic expression of hemochromatosis in 85 homozygous siblings. Medicine (Baltimore) 1986;65:36-56.
29 цит. по: Burke W, Imperiale TF. Hemochromatosis. In: Burke W, Daly M, Garber JE, et al, eds. Genetics in Primary Care. New York: McGraw-Hill, 2008:223-236.
30 цит. по: Cook JD, Skikne BS, Lynch SR, et al. Estimates of iron sufficiency in the US population. Blood 1986;68:726-731.
31 цит. по: Milder MS, Cook JD, Stray S, et al. Idiopathic hemochromatosis, an interim report. Medicine (Baltimore) 1980;59:34-49.
32 цит. по: Adams PC. HFE hemochromatosis is a low penetrance disease. Liver Int 2003;23:407-410.
33 цит. по: Valberg LS, Edwards CQ, Ludwig J, et al. Serum ferritin and body iron stores in familial hemochromatosis. N Engl J Med 1978;299:1058-1062.
34 цит. по: Guyader D, Jacquelinet C, Moirand R, et al. Noninvasive prediction of fibrosis in C282Y homozygous hemochromatosis. Gastroenterology 1998;115:929-936.
35 цит. по: Walsh A, Dixon JL, Ramm GA, et al. Noninvasive markers of liver fibrosis in haemochromatosis. Gut 2004;53:717-725.
36 цит. по: Parkes J, البطن R, Graham-Stewart J, et al. Reliability of serum ferritin as a non-invasive marker of liver fibrosis in haemochromatosis. Gut 2006;55:1629-1633.
37 цит. по: National Institutes of Health. Hemochromatosis. NIH Consens Statement Online 1993 Oct 18-20;11(3):1-12.
38 цит. по: Camaschella C, Roetto A, Deaglio S, et al. Ireg1, a human iron-regulated transporter, is mutated in autosomal dominant hemochromatosis. Cell 1999;98:617-626.
39 цит. по: Adams PC, Speechley M, Kertesz C. Economic analysis of hemochromatosis family screening by HFE genotyping of probands or spouses. Am J Gastroenterol 2003;98:1158-1161.
40 цит. по: Bonkovsky HL, Kowdley KV, Bacon BR, et al. Liver biopsy in patients with hemochromatosis: When is it necessary? J Hepatol 2007;46:758-764.
41 цит. по: Allen KJ, سؤالي J, Gurrin LC, et al. Describing the full phenotypic spectrum of HFE hemochromatosis in a population cohort. Hepatology 2008;47:123-130.
42 цит. по: كيمبيرغ P, Pedersen P, Brøndum-Nielsen K. Survival of C282Y homozygous patients with hemochromatosis in Denmark. Scand J Gastroenterol 2008;43:1118-1124.
43 цит. по: Jensen NM, Fenger M, Hansen TM, et al. Clinical manifestations of HFE hemochromatosis in a Danish population-based cohort. Scand J Gastroenterol 2008;43:1125-1132.
44 цит. по: Cartwright GE, Edwards CQ, Cartwright RE, et al. Hereditary hemochromatosis. Phenotypic expression of the disease. Trans Assoc Am Physicians 1979;92:274-282.
45 цит. по: Barton JC, Acton RT, চানکاکا R, et al. Liver disease in C282Y homozygous hemochromatosis. Results from the Hemochromatosis and Iron Overload Screening (HEIRS) Study. Gastroenterology 2007;133:1072-1079.
46 цит. по: Férec C, Quélin C, Raguenes O, et al. Neonatal screening for hemochromatosis. Ann Med Interne (Paris) 2000;151:71-75.
47 цит. по: Distante S, Fargion S, Colombo I, et al. HFE mutations and iron overload in patients with chronic liver disease. Blood Cells Mol Dis 2004;32:346-351.
48 цит. по: كولمبو I, Piperno A, Mariani R, et al. H63D mutation in hemochromatosis: Clinical relevance and diagnostic implications. Gastroenterology 2000;118:683-689.
49 цит. по: Edwards CQ, Gordeuk VR. HFE hemochromatosis in blacks. Semin Hematol 1998;35:88-94.
50 цит. по: Holtzman NA, Watson MS, eds. Promoting Safe and Effective Genetic Testing in the United States. Baltimore: Johns Hopkins University Press, 1998.
51 цит. по: Grosse SD, Khoury MJ. Genetic tests and public health: A conceptual framework. Am J Epidemiol 2006;163:673-679.
52 цит. по: ماکیلنبرگ JP, Weiss JO, Wertz DC. Genetic discrimination: Perceptions of risk among individuals at increased risk for genetic disorders. Am J Hum Genet 2000;66:960-969.
53 цит. по: Parsons EP, Halliday JL, Lewis B, et al. Psychological impact of genetic screening for hemochromatosis: A randomized controlled trial. Hepatology 2001;33:339-345.
54 цит. по: ماکیلنبرگ JP, Weiss JO, Wertz DC. Genetic discrimination and genetic testing: Perceptions of discrimination by at-risk individuals and the public. Public Health Rep 2001;116:577-587.
55 цит. по: Dixon JL, Williams Y, Scott DF, et al. Acceptability of population screening for haemochromatosis. Med J Aust 2006;185:129-132.
56 цит. по: Tavill AS, Bishop JW, Anderson J, et al. Screening for hemochromatosis: A population-based study. N Engl J Med 2001;344:1954-1959.
57 цит. по: Adams PC, Bradley D, Halverson C, et al. Evaluation of transferrin saturation as a screening test for hemochromatosis. Can J Gastroenterol 2000;14:81-85.