Hereditary angioedema (HAE) presents a significant diagnostic challenge due to its rarity and varied clinical manifestations. This autosomal dominant genetic disorder, stemming from mutations in the C1 esterase inhibitor gene, affects approximately 1 in 50,000 individuals globally. Characterized by episodes of nonpitting angioedema, HAE can impact subcutaneous or submucosal tissues, most commonly affecting the face, limbs, and potentially life-threatening areas like the larynx. Abdominal pain is another frequent and debilitating symptom. Recognizing HAE is crucial as misdiagnosis can lead to ineffective treatments and increased patient morbidity. Understanding the nuances of Hereditary Angioedema Diagnosis is paramount for clinicians to ensure timely and appropriate management, improving patient outcomes and quality of life. This article aims to provide an in-depth exploration of HAE, focusing on its clinical presentation, diagnostic pathways, and the critical role of accurate diagnosis in effective treatment strategies.
Etiology and Pathophysiology of Hereditary Angioedema
Angioedema, broadly defined as swelling resulting from increased vascular permeability, can be categorized into allergic and nonallergic forms. Hereditary angioedema falls under the nonallergic classification, alongside conditions such as renin–angiotensin–aldosterone system blocker-induced angioedema and acquired angioedema. HAE is further sub-divided into three primary types, each sharing similar clinical features but differing in their underlying mechanisms and hereditary angioedema diagnosis criteria.
Type 1 and Type 2 HAE are both attributed to mutations in the SERPING1 gene, responsible for encoding the C1-inhibitor protein. This protein is a crucial serine protease inhibitor that regulates several key biological systems, including the coagulation cascade, contact activation system, fibrinolytic system, and complement system. The distinction between Type 1 and Type 2 HAE lies in the functionality of the C1-inhibitor protein. In Type 1 HAE, which constitutes the majority of cases (80–85%), patients produce insufficient quantities of C1-inhibitor protein. Conversely, Type 2 HAE (15–20% of cases) is characterized by the production of a dysfunctional C1-inhibitor protein, despite having normal or even elevated levels. Both types result from SERPING1 gene mutations, with Type 1 often linked to various mutation types (missense, nonsense, insertions, deletions), while Type 2 is frequently associated with missense mutations in exon 8. For effective physiological function, particularly concerning the contact activation system, a C1-inhibitor protein activity level above 40% is considered necessary. HAE exhibits a dominant-negative inheritance pattern, meaning a mutation in a single allele can disrupt the function of the wild-type allele, leading to disease manifestation even in heterozygotes.
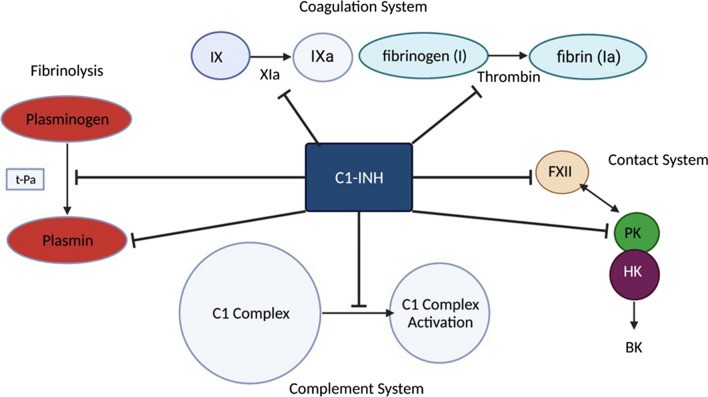 C1 Esterase Inhibitor (C1-INH) Pathways: Illustrating the inhibitory role of C1-INH in fibrinolysis, coagulation, contact, and complement systems, crucial for understanding hereditary angioedema diagnosis and pathophysiology.
C1 Esterase Inhibitor (C1-INH) Pathways: Illustrating the inhibitory role of C1-INH in fibrinolysis, coagulation, contact, and complement systems, crucial for understanding hereditary angioedema diagnosis and pathophysiology.
The primary mediator in HAE pathophysiology is bradykinin. A deficiency or dysfunction of C1-inhibitor protein leads to an overactivation of the contact system, resulting in the excessive production of bradykinin. Bradykinin, in turn, increases vascular permeability, causing the characteristic edema seen in HAE. Furthermore, C1-inhibitor protein normally inhibits plasma kallikrein, an enzyme that converts high-molecular-weight kininogen (HMWK) to bradykinin. Reduced C1-inhibitor function allows plasma kallikrein levels to rise, amplifying bradykinin production. Plasma kallikrein also enhances the active form of factor XII (factor XIIa), creating a positive feedback loop that further promotes bradykinin generation. Bradykinin’s actions on endothelial cells, nociceptors, and bronchial smooth muscle explain the edema, pain, and cough commonly observed in HAE patients. It’s noteworthy that angiotensin-converting enzyme (ACE) degrades bradykinin. Consequently, ACE inhibitors can sometimes induce angioedema (ACE inhibitor-induced angioedema) by preventing bradykinin breakdown.
Type 3 HAE, also referred to as HAE with normal C1-inhibitor activity, is a rarer form and less understood. Initially linked to estrogen dependency, it is now recognized as a heterogeneous group. While elevated estrogen levels can exacerbate symptoms, particularly in women, the underlying mechanisms are more complex. Increased estrogen can lead to higher levels of factor XIIa, contributing to bradykinin overproduction. However, current guidelines recommend classifying HAE with normal C1-inhibitor (HAE-nl-C1-INH) based on specific genetic mutations, including genes like kininogen-1 (HAE-KNG1), plasminogen (PLG-HAE), myoferlin (MYOF-HAE), heparan sulfate-glucosamine 3-sulfotransferase 6 (HS3ST6), factor XII (HAE-F12), and angiopoietin-1 (HAE-ANGPT-1). Therefore, the term “Type 3 HAE” or “HAE-nl-C1-INH” is now considered a broad category encompassing these various mutations rather than a distinct pathological classification. Notably, gain-of-function mutations in the F12 gene, encoding coagulation factor XII, have been identified in some HAE-nl-C1-INH cases, leading to increased bradykinin levels. Accurate hereditary angioedema diagnosis requires consideration of these diverse etiologies and genetic underpinnings.
Clinical Presentation: Key to Hereditary Angioedema Diagnosis
The clinical presentation of hereditary angioedema is highly variable among individuals, making hereditary angioedema diagnosis a clinical challenge. HAE typically manifests as acute, episodic attacks of nonpitting angioedema affecting the skin and submucosal tissues, often accompanied by significant abdominal pain. While angioedema can occur in various body regions, the face and extremities are most commonly affected. Cutaneous angioedema in HAE is characterized by its nonpitting nature and poorly defined borders, distinguishing it from other forms of edema.
Beyond cutaneous manifestations, visceral involvement is a critical aspect of HAE. The gastrointestinal and respiratory tracts are the most frequently affected visceral sites. Gastrointestinal angioedema can cause severe abdominal pain, often described as cramping and debilitating. Laryngeal edema, although less common as an initial symptom, is a life-threatening complication of HAE. It can progress rapidly and lead to airway obstruction and asphyxiation if not promptly recognized and treated. The onset of laryngeal edema can be insidious, with severity increasing over several hours. Therefore, vigilance for respiratory symptoms is crucial in hereditary angioedema diagnosis and management.
Age of onset and symptom duration are also important considerations in hereditary angioedema diagnosis. While HAE can present at any age, most patients become symptomatic before the age of 20. Young children are often asymptomatic, but symptom onset typically occurs during childhood or adolescence, frequently worsening around puberty. However, the absence of early childhood symptoms should not rule out HAE in older patients presenting with characteristic angioedema. Acute HAE attacks typically last for 2 to 5 days and resolve spontaneously without intervention in many cases. This episodic and self-limiting nature of attacks is a key feature distinguishing HAE from chronic angioedema conditions.
Triggers for HAE attacks are diverse and not always identifiable. While some attacks may be preceded by triggers, many occur spontaneously. Common triggers reported by patients include trauma, medical or dental procedures, psychological stress, hormonal changes (e.g., oral contraceptives, menstruation), infections, and ACE inhibitors. However, the response to triggers can vary significantly among individuals, with the same trigger causing different severity of attacks in different patients or even in the same patient at different times. Interestingly, some studies suggest a potential association between HAE and epilepsy, although the underlying mechanisms are still under investigation.
When considering hereditary angioedema diagnosis, it is essential to differentiate HAE from other causes of angioedema, particularly allergic angioedema. The absence of urticaria is a crucial clinical feature that helps distinguish HAE from allergic reactions. Patients experiencing an HAE attack should not exhibit urticaria (hives) on physical examination. Furthermore, the absence of signs of infection, such as warmth, pain, or fever, in the affected area is also important in ruling out infectious causes of swelling. In cases where HAE is suspected, but the diagnosis is uncertain, the administration of an ACE inhibitor might be considered cautiously. ACE inhibitors can exacerbate HAE symptoms, potentially providing further clinical evidence to support the diagnosis in certain cases.
A particular challenge in hereditary angioedema diagnosis is HAE with normal C1-inhibitor levels (HAE-nl-C1-INH). In this subtype, clinical findings are paramount. Patients with HAE-nl-C1-INH present with typical HAE symptoms but have normal C1-inhibitor protein levels and function on standard laboratory tests. Diagnosis in these cases often relies on a combination of clinical presentation, a known F12 gene mutation (in some subtypes), a positive family history of HAE, and documented treatment failure with chronic high-dose antihistamine therapy (which is ineffective in HAE). Therefore, a thorough clinical evaluation is indispensable for accurate hereditary angioedema diagnosis, especially in HAE-nl-C1-INH.
Laboratory Tests in Hereditary Angioedema Diagnosis
Laboratory testing plays a crucial role in confirming hereditary angioedema diagnosis, particularly for Type 1 and Type 2 HAE. The hallmark laboratory findings in HAE are deficiencies in C1 esterase inhibitor, leading to elevated bradykinin levels. However, bradykinin levels are not routinely measured clinically. Instead, diagnostic laboratory tests focus on measuring C1-INH protein levels, C1-INH functional levels, and C4 levels.
Low C1-INH protein levels are characteristic of Type 1 HAE, resulting from mutations that impair protein production. C1-INH functional assays measure the activity of the C1-inhibitor protein, which is reduced in both Type 1 and Type 2 HAE, although the underlying reasons differ. In Type 2 HAE, C1-INH protein levels may be normal or even elevated, but the protein is dysfunctional. Therefore, measuring both C1-INH protein levels and functional activity is essential for differentiating between Type 1 and Type 2 HAE in some cases and confirming hereditary angioedema diagnosis in general.
Measurement of C4 levels is another valuable diagnostic tool in HAE. C4 is a component of the complement system, and its consumption is increased in HAE due to the dysregulation of the complement cascade. Low C4 levels are frequently observed in patients with HAE, particularly during acute attacks. However, C4 levels can fluctuate, and a single measurement may not always be conclusive. While monitoring C4 levels can be helpful, especially during acute episodes, it is important to note that random C4 level testing alone has a sensitivity of approximately 80% for HAE hereditary angioedema diagnosis.
D-dimer levels, markers of coagulation and fibrinolysis activation, may also be elevated during acute HAE attacks. While not specific to HAE, elevated D-dimer levels can provide supportive evidence in the context of suspected acute HAE. In addition to C4 and D-dimer, physicians may also check C1 and/or C3 levels as part of a broader diagnostic workup, although these are less specific for HAE than C4 and C1-INH assays. If any of these laboratory values are significantly below the normal range (e.g., ≤ 50% of normal), repeat testing in 1–3 months is recommended to confirm persistent abnormalities and exclude transient fluctuations due to acute illness. It is crucial to emphasize that a definitive hereditary angioedema diagnosis typically requires both compatible clinical symptoms and supportive laboratory findings.
Genetic testing for SERPING1 gene mutations can also be used to diagnose HAE. Genetic testing can identify specific mutations associated with HAE Type 1 and Type 2. However, genetic testing has limitations in predicting disease severity and progression. The same genetic mutation can manifest differently in different individuals, leading to variable symptom severity and frequency. Furthermore, genetic testing is not always readily available or cost-effective in all clinical settings. Therefore, while genetic testing can be a valuable adjunct in hereditary angioedema diagnosis, it is not always necessary or sufficient for establishing the diagnosis, particularly in resource-limited settings or when clinical and standard laboratory findings are conclusive.
Treatment and Management Strategies for Hereditary Angioedema
Acute Treatment of HAE Attacks
In the absence of treatment, acute HAE attacks typically resolve within 3 to 5 days. However, the potential for life-threatening complications, such as laryngeal edema and severe abdominal pain, necessitates prompt and effective acute treatment. The primary goals of acute treatment are to rapidly alleviate symptoms, prevent attack progression, and avert serious sequelae. Early intervention is crucial, with studies demonstrating better outcomes when treatment is initiated within 6 hours of symptom onset. Therefore, on-demand treatment strategies, allowing patients to self-administer medication at the first sign of an attack, are essential for effective HAE management.
Available acute treatment options include plasma-derived C1-INH (pdC1-INH), recombinant human C1-INH (rhC1-INH), ecallantide, and icatibant. Patient education on self-administration techniques and ensuring access to two home doses of acute medication are vital components of HAE care. Subcutaneous (SC) formulations offer advantages in self-administration compared to intravenous (IV) options. These acute therapies are generally effective within 60 minutes, with symptom relief typically observed within 2 hours. A second dose may be required if symptoms worsen or do not improve adequately. In situations where specific HAE medications are unavailable, fresh frozen plasma (FFP) containing C1-INH can be considered as a last resort. However, FFP is not a preferred treatment due to limited evidence of efficacy and potential for adverse effects. It is crucial to recognize that epinephrine, antihistamines, and glucocorticoids, commonly used for allergic angioedema, are ineffective in treating HAE because HAE is mediated by bradykinin, not histamine. Similarly, ACE inhibitors should be discontinued in HAE patients due to their potential to exacerbate attacks by increasing bradykinin levels.
Intravenous (IV) C1-INH concentrates, pdC1-INH and rhC1-INH, are highly effective acute treatments. IV pdC1-INH is available as plasma-derived concentrates (Cinryze or Berinert). The recommended dose is 20 units/kg, adjusted to vial sizes. Caution is needed during reconstitution to avoid shaking, which can denature the protein. Headache is a common side effect, and thrombotic events have been reported rarely. RhC1-INH is derived from transgenic rabbit milk. Rabbit allergy is a contraindication. While effective, rhC1-INH has a shorter half-life than pdC1-INH, requiring higher doses for acute treatment and limiting its use for prophylaxis. The recommended dose is 50 units/kg, up to a maximum of 4200 units per dose. Both IV C1-INH products are approved for use in children aged 5 years and older.
Icatibant is a bradykinin B2-receptor antagonist administered by subcutaneous injection. It is approved for patients aged 18 years and older in the United States. Dosing is weight-based, ranging from 10 mg to 30 mg per dose. A second dose can be given after 6 hours if symptoms persist, with a maximum of three doses in 24 hours. Caution is advised in patients with angina or coronary artery disease, as icatibant may reduce coronary blood flow.
Ecallantide is another subcutaneous option, a recombinant plasma kallikrein inhibitor. It blocks bradykinin production. Approved by the FDA for treating HAE attacks in patients aged 12 years and older in the U.S., ecallantide carries a risk of anaphylaxis and allergic reactions. Therefore, it should only be administered in healthcare settings equipped to manage anaphylaxis. The adult dose is 30 mg, administered as three separate 10 mg subcutaneous injections in the abdomen, upper arm, or thigh, away from the angioedema site. A second 30 mg dose can be given as early as 1 hour after the first dose if needed.
Laryngeal attacks are associated with significantly increased mortality, requiring immediate emergency care. Patients with laryngeal, throat, or tongue swelling should seek emergency medical attention immediately. Elective intubation should be considered for patients with respiratory distress despite initial treatment. In cases of failed intubation, emergent cricothyrotomy may be necessary to secure the airway. Gastrointestinal attacks, while often severe, usually resolve without specific intervention beyond on-demand HAE medication. Symptomatic treatment with rehydration, antiemetics, antidiarrheals, or laxatives may be helpful in managing gastrointestinal symptoms.
Short-Term Prophylaxis for HAE
Short-term prophylaxis is indicated to prevent HAE attacks in at-risk situations, such as invasive procedures or anticipated stressful events. Trauma and stress are known triggers for HAE attacks. Procedures requiring short-term prophylaxis include major dental work, oral surgery, endotracheal intubation, and endoscopies. For patients already on long-term prophylaxis, the dosing schedule of their prophylactic medication can be adjusted to provide additional protection before the procedure or event. Alternatively, a short course of androgens, initiated 5 days prior to the procedure and continued for 2–3 days afterwards, can be used. If pdC1-INH is used for short-term prophylaxis, the standard acute treatment dose should be administered 1 to 12 hours before the procedure. Ecallantide and icatibant are not suitable for prophylaxis due to their short half-lives. Tranexamic acid (TXA), an antifibrinolytic agent, is sometimes used for short-term prophylaxis, although its efficacy is debated. TXA can reduce bradykinin levels by interfering with plasmin activity. A 2021 case report suggests that tranexamic acid may be particularly useful for short-term prophylaxis in ACE inhibitor-induced angioedema.
Long-Term Prophylaxis for HAE
The decision to initiate long-term prophylaxis for HAE is individualized and depends on factors such as attack frequency, severity, impact on quality of life, access to acute treatment, and comorbid conditions. First-line long-term prophylactic therapies include lanadelumab (a monoclonal plasma kallikrein inhibitor), IV pdC1-INH (Cinryze), and SC pdC1-INH (Haegarda). Anabolic androgens or antifibrinolytics are considered second-line options due to their side effect profiles.
Cinryze, an IV pdC1-INH product, is approved for long-term prophylaxis in children and adults. It is administered as an infusion every 3 to 4 days, with doses of 1000 or 2500 units depending on age. Long-term IV access poses challenges, including infection risk and vein damage. Indwelling ports are generally not recommended due to infection risks. Haegarda, a subcutaneous pdC1-INH formulation, offers a first-line alternative that avoids IV access risks. It is administered subcutaneously every 3 to 4 days at a dose of 60 units/kg. PdC1-INH products are the preferred option for long-term prophylaxis in pregnant and breastfeeding women.
Lanadelumab, a monoclonal antibody targeting plasma kallikrein, was FDA-approved in 2020 for patients aged 12 years and older. Its long duration of action allows for less frequent dosing, typically 300 mg subcutaneously every two weeks. After 6 months of therapy with good attack control, the dosing interval can be extended to 300 mg every 4 weeks.
Berotralstat is a recently approved oral plasma kallikrein inhibitor for patients aged 12 years and older. It is administered once daily at a dose of 150 mg with food. A 110 mg dose is available for patients taking strong inhibitors of BCRP or P-gp transporters. A primary concern with berotralstat is the potential for QT prolongation, requiring monitoring. Long-term safety and efficacy data are still being collected.
Danazol, an oral anabolic androgen, is a widely available option for long-term prophylaxis in the US. While effective, it has dose-related side effects and is considered a second-line agent. The lowest effective dose should be used, typically ranging from 50 to 200 mg daily or every other day. Two dosing strategies can be used: starting with a high dose (400–600 mg daily) and tapering down or starting with a low dose (100 mg daily) and titrating up as needed. Androgens should be avoided in prepubertal children and pregnant women.
Tranexamic acid (TXA), an antifibrinolytic, is another second-line, off-label option for long-term prophylaxis. It is often preferred over androgens in pregnant women and children when first-line options are not suitable. Typical dosing starts at 500 mg orally two to three times daily, titrated up to a maximum of 3 g daily as tolerated. The risk of thrombosis should be monitored during TXA therapy.
Conclusion: The Importance of Accurate Hereditary Angioedema Diagnosis
Prompt recognition and appropriate treatment of hereditary angioedema are crucial to prevent complications associated with delayed or inaccurate hereditary angioedema diagnosis. HAE commonly presents with acute episodes of nonpitting angioedema affecting subcutaneous or submucosal tissues, often in the face and extremities. Abdominal pain and potentially life-threatening laryngeal edema are significant associated symptoms. Distinguishing HAE from allergic angioedema by the absence of urticaria is essential for appropriate management. Furthermore, patients with HAE should not exhibit signs of infection or fever during an attack. While young children with HAE are often asymptomatic, symptom onset typically occurs before age 20. Laboratory testing, including C1-INH protein levels, C1-INH functional levels, and C4 levels, is critical for confirming hereditary angioedema diagnosis.
Effective acute treatment is paramount in preventing attack progression, with medications like pdC1-INH, rhC1-INH, ecallantide, and icatibant providing rapid relief. Short-term and long-term prophylaxis strategies are available to reduce the frequency and severity of HAE attacks. Long-term prophylaxis first-line therapies include IV pdC1-INH (Cinryze), SC pdC1-INH (Haegarda), and lanadelumab. Second-line options include anabolic androgens and antifibrinolytics. Haegarda offers a subcutaneous option for prophylaxis, avoiding IV access complications. Lanadelumab, with its less frequent dosing schedule, and berotralstat, an oral option, represent newer advances in HAE management. Danazol, while effective, is a second-line option due to side effects, with dosing individualized to minimize adverse effects. Tranexamic acid is another second-line option, particularly useful in pregnant women and children, but requires monitoring for thrombosis risk. Accurate and timely hereditary angioedema diagnosis, coupled with appropriate treatment strategies, significantly improves the lives of individuals affected by this rare and potentially debilitating condition.
Acknowledgements
Funding
No funding or sponsorship was received for this study or publication of this article.
Author Contributions
Study concept and design: Evan S. Sinnathamby, Peter P. Issa, Logan Roberts, Haley Norwood, Kevin Malone, Harshitha Vemulapalli, Shahab Ahmadzadeh, Elyse M. Cornett, Sahar Shekoohi, Alan D. Kaye. Analysis and interpretation of data: Evan S. Sinnathamby, Peter P. Issa, Logan Roberts, Haley Norwood, Kevin Malone, Harshitha Vemulapalli, Shahab Ahmadzadeh, Elyse M. Cornett, Sahar Shekoohi, Alan D. Kaye. Drafting of the manuscript: Evan S. Sinnathamby, Peter P. Issa, Logan Roberts, Haley Norwood, Kevin Malone, Harshitha Vemulapalli, Shahab Ahmadzadeh, Elyse M. Cornett, Sahar Shekoohi, Alan D. Kaye critical revision of the manuscript for important intellectual content: Evan S. Sinnathamby, Peter P. Issa, Logan Roberts, Haley Norwood, Kevin Malone, Harshitha Vemulapalli, Shahab Ahmadzadeh, Elyse M. Cornett, Sahar Shekoohi, Alan D. Kaye statistical analysis: Evan S. Sinnathamby, Peter P. Issa, Logan Roberts, Haley Norwood, Kevin Malone, Harshitha Vemulapalli, Shahab Ahmadzadeh, Elyse M. Cornett, Sahar Shekoohi, Alan D. Kaye.
Compliance with Ethics Guidelines
This article is based on previously conducted studies and does not contain any new studies with human participants or animals performed by any of the authors.
Disclosures
Evan S. Sinnathamby has nothing to disclose. Peter P. Issa has nothing to disclose. Logan Roberts has nothing to disclose. Haley Norwood has nothing to disclose. Kevin Malone has nothing to disclose. Harshitha Vemulapalli has nothing to disclose. Shahab Ahmadzadeh has nothing to disclose. Elyse M. Cornett has nothing to disclose. Sahar Shekoohi has nothing to disclose. Alan D. Kaye has nothing to disclose.
Data Availability
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
Contributor Information
Evan S. Sinnathamby, Email: [email protected]
Peter P. Issa, Email: [email protected]
Logan Roberts, Email: [email protected].
Haley Norwood, Email: [email protected].
Kevin Malone, Email: [email protected].
Harshitha Vemulapalli, Email: [email protected].
Shahab Ahmadzadeh, Email: [email protected].
Elyse M. Cornett, Email: [email protected]
Sahar Shekoohi, Email: [email protected].
Alan D. Kaye, Email: [email protected]
References
Associated Data
Data Availability Statement
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.