Introduction
IgG4-Related Disease (IgG4-RD) is a relatively recently identified condition that is being diagnosed with increasing frequency. This rise in diagnosis is largely due to greater awareness among clinicians. IgG4-RD can affect both men and women and has been observed in a wide range of organs. The salivary and lacrimal glands, pancreas, and liver are among the most commonly involved. Understanding the nuances of Igg4 Diagnosis is crucial for effective patient management.
Areas Covered
Diagnosing and managing IgG4-RD remains complex. Researchers are actively investigating suitable biomarkers and effective therapies. Currently, histological examination of affected tissues remains the definitive method for confirming a diagnosis. However, it’s important to note that serum IgG4 levels are not always reliable for diagnosis, being neither consistently elevated in all cases nor specific to IgG4-RD. Glucocorticoids are still considered the most effective initial treatment to manage the condition, but there is ongoing research to determine the best approaches for long-term maintenance therapy. This review provides an updated overview of current strategies for igg4 diagnosis and management of IgG4-RD. Much of our current knowledge is based on retrospective studies and expert consensus guidelines. However, ongoing prospective studies, clinical trials, and a deeper understanding of the disease’s underlying mechanisms offer promising avenues for advancement in this field.
Keywords: IgG4-RD, IgG4, Diagnosis and Management, plasmablasts, rituximab, glucocorticoids, autoimmune
1. INTRODUCTION
IgG4-related disease (IgG4-RD) is characterized as a systemic immune-mediated condition causing fibro-inflammation. It frequently affects multiple organ systems, either at the time of initial diagnosis or as the disease progresses. The concept of IgG4-RD is relatively new, originating from observations of autoimmune pancreatitis (AIP) about a decade ago. Dr. Kamisawa, a gastroenterologist in Japan, noted similar pathological features in other organs affected in patients initially diagnosed with AIP [1]. It is now recognized that IgG4-RD can impact virtually any organ system, with the pancreas and biliary system, along with salivary and lacrimal glands, being most frequently reported [2]. Igg4 diagnosis can be challenging because organ involvement can occur at different times, either simultaneously or sequentially. The disease is characterized by lesions that resemble tumors, containing a high density of IgG4-producing plasma cells, lymphocytes, and fibrosis. This can mimic malignancy. Importantly, IgG4-RD is responsive to glucocorticoid treatment, and prompt and accurate igg4 diagnosis can prevent significant complications and long-term organ damage. A range of conditions previously considered separate idiopathic fibrosing disorders, such as Ormond’s disease (idiopathic retroperitoneal fibrosis), Riedel’s thyroiditis, sclerosing mesenteritis, and Mikulicz disease, are now recognized as manifestations within the IgG4-RD spectrum.
Misdiagnosis of IgG4-related diseases is common. This is partly due to a lack of familiarity with the condition among clinicians and partly due to the absence of a consistently reliable biomarker. Elevated serum IgG4 concentrations [3] are often considered a key biomarker for igg4 diagnosis and differentiation from conditions that mimic IgG4-RD [4], as listed in Table 1. However, relying solely on serum IgG4 levels can be misleading. The cornerstone of accurate igg4 diagnosis for this multifaceted condition remains clinicohistopathological confirmation.
Table 1. Mimickers of IgG4-RD
| Autoimmune | Malignancy | Other |
|---|---|---|
| Antineutrophil cytoplasmic antibody-associated vasculitis | Adenocarcinoma and Squamous Cell Carcinoma | Castleman’s Disease |
| Granulomatosis with Polyangitis | Extranodal Marginal Zone lymphoma | Cutaneous Plasmacytosis |
| Eosinophilic Granulomatosis with Polyangitis | Inflammatory myofibroblastic tumor | Erdheim-Chester Disease |
| Microscopic Polyangitis | Lymphoplasmocytic Lymphoma | Inflammatory Bowel Disease |
| Sarcoidosis | Lymphoproliferative Disease | Perforating Collagenosis |
| Sjogren’s Disease | Follicular Lymphoma | Primary Sclerosing Cholangitis |
| Rhinosinusitis | ||
| Rosai-Dorfman Disease | ||
| Splenic Sclerosing Angiomatoid Nodular transformation | ||
| Xanthogranuloma |
The epidemiology of IgG4-RD is not yet fully understood due to its rarity and frequent misdiagnosis. A study in Japan in 2009 [5] reported a prevalence of 8000 cases in their population, with a higher incidence in males and an average age of onset of 58.8 years, peaking between 61 and 70 years. Studies in Western countries have shown similar age and sex distributions. However, accurate estimates of incidence and prevalence remain unclear in the United States and other regions, hindering efforts to identify at-risk populations [6]. While IgG4-RD is more commonly seen in older men, head and neck involvement appears to be equally prevalent in both genders [7]. Although typically reported in older adults, cases have been documented in children, predominantly females [8]. Further epidemiological research is needed to better define the global burden of IgG4-RD and improve igg4 diagnosis rates across diverse populations.
2. CLINICAL PRESENTATION
Patients with IgG4-RD typically present with the subacute development of a mass lesion within an organ. The disease itself is generally not associated with systemic symptoms or pain. Patients often seek medical attention due to cosmetic concerns from salivary or lacrimal gland enlargement, obstructive symptoms caused by mass effect (e.g., obstructive jaundice in pancreatitis and cholangitis), or obstructive uropathy due to retroperitoneal fibrosis.
The frequency of organ involvement and the initial symptoms of IgG4-RD can vary significantly depending on the medical specialty evaluating the patient. Gastroenterologists frequently see autoimmune pancreatitis and cholangitis as primary presentations, while rheumatologists may more often encounter salivary and lacrimal gland involvement. Accurate igg4 diagnosis requires considering the diverse range of potential clinical presentations.
Approximately 60% of patients with IgG4-RD have involvement of more than one organ at the time of diagnosis. A detailed medical history and physical examination are crucial for identifying additional, potentially asymptomatic, organ involvement. For example, patients with obstructive jaundice due to autoimmune pancreatitis may also have bilateral submandibular gland involvement. This finding can raise clinical suspicion for IgG4-RD, potentially averting unnecessary invasive procedures like pancreatectomy when cancer is initially suspected. Many other organ involvements can be asymptomatic and may only be detected through comprehensive imaging of the chest and abdomen. Therefore, thorough investigation is vital for accurate igg4 diagnosis and staging.
Lymphadenopathy has been reported in 25–60% of IgG4-RD patient cohorts [6, 7], making lymphoproliferative disorders a significant diagnostic mimic. Lymph node biopsies are not typically diagnostic for IgG4-RD because the characteristic fibrosis is usually absent in this tissue. Other histological features, including IgG4 lymphoplasmacytic infiltrates, are non-specific in lymph nodes. While five histological subtypes of IgG4-related lymphadenopathy have been proposed [9, 10], none are specific for igg4 diagnosis. However, lymph node biopsy is essential to rule out malignancy, particularly lymphoma, in the differential diagnosis.
A history of allergies, including asthma, eczema, seasonal allergies, or food allergies, is present in approximately 30–40% of patients with IgG4-RD [11]. Further research is necessary to clarify if and how allergic predisposition contributes to the pathogenesis of IgG4-RD. Understanding these potential associations could refine igg4 diagnosis and risk stratification.
3. PATHOGENESIS
The precise mechanisms underlying IgG4-RD pathogenesis are not fully understood. Over recent years, several models have been proposed to explain disease development. The abundance of plasma cells in affected tissues and elevated IgG4 antibody levels suggest a central role for B cells and plasma cells. This is supported by the observed clinical improvement in IgG4-RD patients following B cell depletion therapies. Recent studies suggest that dysregulation of follicular helper T cells and CD4+ cytotoxic T cells may orchestrate IgG4-RD pathogenesis. These T cell subsets may contribute by secreting pro-fibrotic cytokines and by inducing IgG4 class switching, plasmablast expansion, and autoantibody production [12, 13, 14, 15]. Innate immunity also appears to play a role, with studies showing that innate immune cells like plasmacytoid dendritic cells and monocytes isolated from IgG4-RD patients can promote IgG4 production [16]. Further research into these complex immune interactions is crucial for advancing both igg4 diagnosis and targeted therapies.
The pathogenic role of the IgG4 molecule itself in IgG4-RD is not fully established. IgG4 antibodies are generally considered non-inflammatory and are thought to dampen immune system activation in most contexts. Pathogenic roles for IgG4 are established in only a few specific conditions, such as pemphigus vulgaris and thrombotic thrombocytopenic purpura. In IgG4-RD, correlations between serum IgG4 levels and disease severity, as well as the detection of IgG4 immune complexes in IgG4-related tubulointerstitial nephritis, hint at a potential pathogenic role for this antibody. However, this requires further investigation to be confirmed. Current igg4 diagnosis strategies rely on broader clinicopathological features rather than solely on the presumed pathogenicity of IgG4 itself.
4. DIAGNOSIS
The most reliable approach to igg4 diagnosis integrates clinical history, thorough physical examination, laboratory testing, imaging, and, critically, histopathological examination. Histopathologic confirmation is particularly important in cases presenting with tumor-like lesions to exclude malignancy. IgG4-RD should be considered when mass lesions are found in typical organs such as the pancreas, salivary and lacrimal glands, lungs, liver, kidneys, and retroperitoneum. A crucial initial step is to rigorously exclude conditions that mimic IgG4-RD. While IgG4-RD may have been historically under-recognized, it is essential to include it in the differential diagnosis when evaluating patients with suggestive clinical findings. It is paramount to conduct a thorough workup to ensure that malignancy or infection is not the underlying cause of fibro-inflammatory organ involvement. Accurate igg4 diagnosis depends on a systematic and comprehensive evaluation.
Several conditions previously categorized as idiopathic fibrosing diseases are now understood to be manifestations of IgG4-RD. Table 2 lists conditions once considered separate entities that are now largely recognized as different presentations of IgG4-RD [17–21]. This broader understanding of IgG4-RD is essential for accurate igg4 diagnosis and classification.
Table 2. Conditions previously known as separate entities now mostly recognized as part of IgG4-RD
| Mikulicz’s disease |
|---|
| Kuttner’s tumor |
| Eosinophilic angiocentric fibrosis |
| Riedel’s thyroiditis |
| Idiopathic cervical fibrosis |
| Pulmonary inflammatory pseudotumor |
| Chronic sclerosing aortitis |
| Preaortitis and periarteritis |
| Autoimmune pancreatitis |
| Sclerosing cholangitis |
| Idiopathic tubulointerstitial nephritis |
| Retroperitoneal fibrosis |
| Sclerosing mesenteritis |
| Inflammatory aortic aneurysm |
| Multifocal fibrosclerosis |
While various diagnostic criteria have been proposed for IgG4-RD, AIP, and IgG4-related kidney disease [22–24], no diagnostic criteria can replace comprehensive clinicopathological correlation for clinical igg4 diagnosis.
1. Laboratory
Initially, elevated serum IgG4 levels were considered central to igg4 diagnosis. However, it is now clear that elevated serum IgG4 is neither necessary nor sufficient for diagnosing IgG4-RD. Studies have demonstrated that elevated IgG4 levels do not definitively confirm IgG4-RD, and normal serum IgG4 levels do not exclude it. Conditions that mimic IgG4-RD, such as pancreatic adenocarcinoma, lymphoma, and ANCA-associated vasculitis, can also present with elevated serum IgG4 levels. Conversely, patients with biopsy-confirmed IgG4-RD can have normal IgG4 concentrations. A meta-analysis of predominantly Caucasian and Asian patients found no specific cut-off value for serum IgG4 levels to definitively diagnose IgG4-RD, although it did show statistically significant pooled sensitivity and specificity for serum IgG4 level testing [25]. However, another study reported the specificity and positive predictive value of elevated serum IgG4 concentrations to be only 60% and 34%, respectively [26]. Therefore, while elevated serum IgG4 can raise suspicion, it is not a standalone marker for igg4 diagnosis.
Typically, an elevated serum IgG4 concentration in the appropriate clinical context, with typical organ involvement, can increase clinical suspicion for IgG4-RD. It is crucial to recognize that elevated IgG4 levels can be found in other conditions, particularly other autoimmune diseases, asthma, allergies, plasma cell dyscrasias, multicentric Castleman disease, and certain malignancies. Patients with multi-organ IgG4-RD involvement are more likely to have higher serum IgG4 concentrations, and normal serum IgG4 is less common in cases with multiple organ involvement. Furthermore, the degree of serum IgG4 elevation correlates with the likelihood of IgG4-RD; very high levels (>5 times the upper limit of normal) are less frequently seen in other conditions. Increased ratios of IgG4 to total IgG (>10%) can also be a helpful finding and may improve diagnostic specificity, particularly when IgG4 concentrations are only mildly elevated [27]. These nuances are important for refining igg4 diagnosis using serological markers.
Monitoring serum IgG4 concentrations may be useful in assessing disease activity in some patients but should not be the sole determinant of disease activity or the need for treatment. Serum IgG4 levels typically decrease rapidly after treatment with glucocorticoids or B cell depletion in most patients, but many patients do not achieve normal levels even during clinical remission. Disease relapses can also occur in approximately 10% of patients with low serum IgG4 levels [28]. In a prospective trial of rituximab in IgG4-RD, higher baseline elevations in serum IgG4, IgE, and blood eosinophil concentrations were predictive of a greater risk of relapse and shorter time to relapse [29], suggesting the value of measuring these parameters before treatment initiation and during follow-up. These factors should be considered in the context of igg4 diagnosis and management.
When interpreting low serum IgG4 levels, it is essential to rule out the prozone phenomenon in the immunoassay. This phenomenon occurs when an excess of antigen interferes with antibody binding, leading to the resolubilization of immune complexes and a falsely low signal. This is a critical consideration because clinicians who rely heavily on elevated serum IgG4 for igg4 diagnosis may miss cases of true IgG4-RD due to inaccurately low readings [30]. Egner et al. found that even four years after the initial report of the prozone phenomenon in IgG4 assays, this error still occurred in 41% of high serum IgG4 measurements, leading to spurious results in their UK cohort [31]. It is strongly recommended to dilute the test sample to ensure antibody concentration remains in excess of antigen and to check for prozoning in the initial sample. This simple step can help identify elevated serum IgG4 concentrations that might otherwise be missed, improving the accuracy of igg4 diagnosis.
Flow cytometry identification of high numbers of plasmablasts in peripheral blood has been proposed as a more sensitive biomarker for disease activity in IgG4-RD than serum IgG4 concentrations. Plasmablasts are typically elevated in active IgG4-RD, even in patients with normal serum IgG4 levels, and have been shown to decrease after rituximab treatment compared to controls [32]. However, plasmablast measurement by flow cytometry on fresh blood samples is technically demanding and not routinely available outside of research settings. Further development of accessible plasmablast assays could enhance igg4 diagnosis and disease monitoring.
Doorenspleet et al. [33] have recently demonstrated the potential of measuring dominant IgG4 B cell receptor clones as a biomarker for IgG4-related cholangitis. Their initial study identified an abundance of these specific clones, measured by next-generation sequencing, in both peripheral blood and tissue of patients with IgG4-related cholangitis [34]. They developed a more practical and cost-effective quantitative polymerase chain reaction (qPCR) technique to measure the ratio of IgG4/IgG RNA of the same B cell receptors. This qPCR assay showed a sensitivity of 94% and specificity of 99% for igg4 diagnosis of IgG4-related cholangitis. In the same study, serum IgG4 concentration showed a sensitivity of 86% and specificity of 73% [33]. While promising, the IgG4/IgG RNA ratio by qPCR requires further validation in larger, independent cohorts to confirm its clinical utility in igg4 diagnosis and management.
Hypocomplementemia has been observed in active IgG4-RD patients, particularly those with systemic disease and renal involvement [35]. However, IgG4 is not traditionally considered a strong complement-binding antibody. The low complement levels associated with renal disease in these patients may be due to higher concentrations of other IgG subclasses, such as IgG1, which are known to activate complement more effectively [36, 37]. Research on anti-hinge antibodies, found in patients with rheumatoid arthritis, may provide further insights into the role of complement activation in IgG4-RD [38]. The role of complement in IgG4-RD pathogenesis and its potential diagnostic utility require further investigation to refine igg4 diagnosis and understand disease mechanisms.
Elevated IgE levels and peripheral eosinophilia are also common features in IgG4-RD, as are low-titer positive antinuclear antibodies (ANA) and rheumatoid factor (RF) [35, 39]. C-reactive protein (CRP) and erythrocyte sedimentation rate (ESR) are non-specific markers of inflammation and are typically not elevated in IgG4-RD unless another inflammatory process is concurrently present, such as in cases of aortitis or cholangitis [35]. There has been some debate among researchers about whether elevated CRP is more suggestive of multicentric Castleman disease rather than IgG4-RD. Currently, it is prudent to consider other diagnoses, including multicentric Castleman disease, if there is significant CRP elevation, and to integrate this laboratory finding with other clinicopathological features for accurate igg4 diagnosis. Table 3 summarizes the most common laboratory findings associated with IgG4-RD.
Table 3. Most common laboratory findings associated with IgG4-RD
| Labs associated with IgG4-RD |
|---|
| IgG4 level > 135 mg/dL |
| Elevated IgG4: IgG ratio >10% |
| Peripheral eosinophilia |
| CRP normal |
| ESR normal |
| ANA low titer positive |
| Elevated IgE levels |
| Hypocomplementemia |
| Increased number of circulating plasmablasts by flow cytometry |
| qPCR of IgG4/IgG RNA |
2. Imaging
Patients with IgG4-RD often present with tumor-like lesions and may have already undergone computed tomography (CT) or magnetic resonance imaging (MRI) of the affected area for initial characterization. Imaging plays a crucial role in igg4 diagnosis and assessing disease extent.
Most studies report that 56–60% of patients with IgG4-RD have multi-organ involvement at the time of diagnosis [40]. However, initial clinical findings may suggest single-organ disease. Therefore, imaging studies are essential to detect asymptomatic organ involvement.
Since many symptoms of IgG4-RD arise from mass effect or organ damage due to fibroinflammatory infiltration, cross-sectional imaging can reveal evidence of additional organ involvement even in asymptomatic patients. Routine imaging of the chest and abdomen is recommended for comprehensive disease assessment at diagnosis. The optimal imaging modality for evaluating disease activity in IgG4-RD is a topic of ongoing discussion. 18-Fluorodeoxyglucose positron emission tomography (FDG PET/CT) has demonstrated diagnostic utility in IgG4-RD due to its ability to identify metabolically active inflammatory lesions and delineate disease extent [41]. However, FDG PET/CT can be expensive and less readily available for routine igg4 diagnosis and monitoring. The consensus is that whole-body imaging is necessary for IgG4-RD assessment, but the choice between contrast-enhanced CT, MRI, or PET/CT can be left to the clinician’s discretion based on availability and clinical context. Certain imaging patterns are highly suggestive of IgG4-RD [41, 42] and are listed in Table 4. Recognizing these patterns aids in igg4 diagnosis.
Table 4. Imaging findings strongly suggestive of IgG4-RD
| Imaging patterns strongly suggestive of IgG4-RD |
|---|
| Symmetric bilateral enlargement of salivary glands with even distribution of 18F-FDG uptake without signs of infection |
| Enlargement of lacrimal glands; Involvement of trigeminal nerve branches; Orbital myositis; Orbital adnexal soft tissue |
| Bronchovascular and septal thickening in the lungs; Pleural or parenchymal nodules |
| Diffusely enlarged pancreas with loss of lobulation with or without pancreaticobiliary duct narrowing; Enhancement around the pancreatic rim; Pancreatic atrophy due to damage |
| Patchy or diffuse thickness of aorta wall and soft tissue around the abdominal aorta |
| Patchy retroperitoneal lesion with moderate to intense 18F-FDG uptake |
| Bilateral renal cortex low density areas |
Gallium SPECT/CT has also been suggested as a potential imaging modality for igg4 diagnosis and treatment response assessment [43]. Being less expensive and more accessible than FDG PET/CT, gallium SPECT/CT could be a valuable alternative if its diagnostic efficacy is confirmed to be comparable [44]. Some evidence suggests that gallium SPECT/CT may even be superior to PET/CT for evaluating renal involvement in IgG4-RD, potentially differentiating renal parenchymal disease from perirenal pseudotumors [45]. Further comparative studies are needed to clarify the optimal role of different imaging modalities in igg4 diagnosis and management.
3. Histopathology
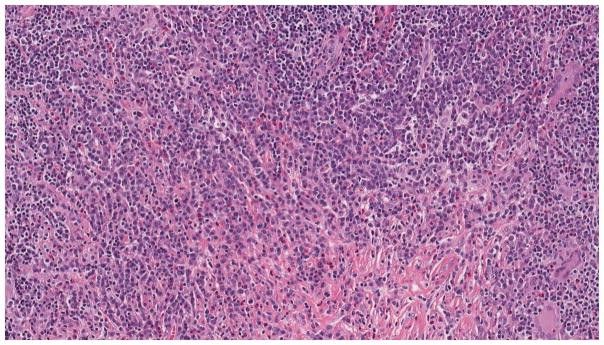
Histopathologic assessment of affected organs remains the gold standard for igg4 diagnosis. Biopsy is crucial for excluding conditions that mimic IgG4-RD, especially malignancy and infection. Needle biopsies may provide sufficient tissue to rule out mimics but may not always yield enough tissue to confidently assess the characteristic histological features of IgG4-RD. The three hallmark histological features of IgG4-RD in tissue are: dense lymphoplasmacytic infiltration, storiform fibrosis (an irregularly whorled fibrotic pattern), and obliterative phlebitis [Figure 1A] [46]. According to consensus guidelines on the pathology of IgG4-RD, the presence of at least two out of these three histological features is typically required for igg4 diagnosis [47]. Additional findings, such as tissue eosinophilia and germinal centers in affected organs, can further strengthen suspicion for IgG4-RD. Conversely, the presence of granulomas, prominent neutrophilic infiltration, or necrosis should raise suspicion for alternative diagnoses, such as infections, sarcoidosis, or granulomatosis with polyangiitis (GPA), and argue against igg4 diagnosis.
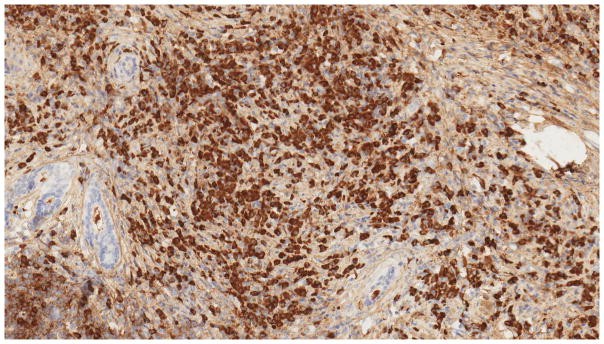
For a definitive pathological igg4 diagnosis, in addition to the characteristic histological features, the presence of IgG4+ plasma cells infiltrating the tissue is necessary [Figure 1B]. The required number of IgG4+ plasma cells and the IgG4+/IgG+ plasma cell ratio have been subjects of debate and can vary across different organs. Consensus pathology guidelines suggest thresholds of at least >10 IgG4+ plasma cells per high-power field (HPF) in some organs, including the pancreas, and >50 IgG4+ plasma cells/HPF in most other organs, along with an IgG4+/IgG+ plasma cell ratio of >40% [47]. These criteria have been associated with a sensitivity of 94.4% and a specificity of 85.7% for igg4 diagnosis [48].
Figure 1. Pathology of a lacrimal gland in a patient with multi systemic IgG4-RD.
Figure 1A: Histopathology of IgG4-RD in a lacrimal gland. High power view showing lymphoplasmacytic infiltration and fibrosis (Hematoxylin and Eosin stain).
Figure 1B: Immunohistochemistry for IgG4 in IgG4-RD. IgG4 staining showing marked infiltration of IgG4+ plasma cells in the lacrimal gland.
However, IgG4+ plasma cell infiltration is not specific to IgG4-RD and can be observed in mimickers of the disease. Therefore, relying solely on the number of IgG4+ plasma cells, without the characteristic histological features, is insufficient for igg4 diagnosis. Over-reliance on IgG4+ plasma cell counts can lead to misdiagnosis in cases of pancreatic cancer, Castleman disease, GPA, sarcoidosis, infections, and lymphomas. While pathology is central to igg4 diagnosis, similar pathological findings in certain tissues without typical organ involvement may be non-specific. Skin, lymph nodes, thyroid, and nasosinus cavities are sites that can show IgG4+ plasma cells in the context of fibroinflammation without necessarily indicating IgG4-RD.
It is also important to acknowledge that a negative or inconclusive biopsy result does not definitively rule out IgG4-RD. This can be due to sampling variability inherent in needle biopsies or because the biopsy may sample a fibrotic area of the lesion with reduced inflammation and fewer IgG4+ cells. In cases of high clinical suspicion, repeat biopsy or consideration of alternative diagnostic approaches may be necessary for accurate igg4 diagnosis.
4. TREATMENT & MANAGEMENT
Currently, there are no randomized controlled trials guiding the treatment of IgG4-RD. Therefore, the optimal evidence-based treatment remains undefined. However, observational studies have demonstrated the effectiveness of certain medications in inducing remission and reducing relapse rates. Treatment choices for IgG4-RD can vary across countries, medical specialties, and based on the specific organs involved [49].
1. Glucocorticoid treatment
Glucocorticoids are the cornerstone of initial therapy for IgG4-RD. Reported response rates to glucocorticoid therapy range from 97–100% [37]. Response to glucocorticoids is even part of the diagnostic criteria for AIP [23]. In most studies, glucocorticoids are used for initial induction therapy, and many clinicians use low-dose prednisone (5–7.5 mg equivalent) for maintenance. Despite initial success, relapse rates are significant. Between 40–76% of patients successfully treated with glucocorticoids will relapse, either in the same organ or a different one, requiring repeated induction treatments and prolonged tapers [50]. Patients with IgG4-RD are often older and at higher risk of developing diabetes due to pancreatic involvement. They are also susceptible to steroid-related complications, including hyperglycemia, cataracts, osteoporosis, and increased cardiovascular risk. International consensus guidelines recommend initial induction therapy with prednisone equivalent at 0.6 mg/kg or 30 mg/day for 2–4 weeks, followed by tapering over 8–12 weeks [4]. Relapses are common during steroid tapering [51]. Relapse episodes are typically treated with the same induction regimen, and many clinicians consider initiating steroid-sparing agents after the first relapse. Some clinicians maintain patients on long-term low-dose steroids for maintenance therapy, but this approach needs to be carefully weighed against the risks of chronic steroid use.
2. Steroid Sparing Strategies
Due to limited evidence on steroid-sparing agents in IgG4-RD and the absence of prospective controlled trials, choosing a specific steroid-sparing strategy is challenging. A recent meta-analysis [49] reported that azathioprine was used in 85% of cases, followed by mycophenolate mofetil, methotrexate, tacrolimus, leflunomide, and cyclophosphamide. Relapses have been reported in patients on low doses of azathioprine (50 mg daily) or mycophenolate (1 g daily). Increasing the dose has been reported to control the disease in some cases, but most responses were achieved in combination with glucocorticoids, making it difficult to assess the efficacy of steroid-sparing agents alone [49]. No studies have directly compared the efficacy of these conventional steroid-sparing agents in IgG4-RD. The choice of immunosuppressive agent is often influenced by the specific organ involvement and the clinician’s specialty. Azathioprine is frequently used as a steroid-sparing agent, particularly in AIP, as it is a commonly used immunosuppressant by gastroenterologists. Further research is needed to establish evidence-based steroid-sparing strategies in IgG4-RD management.
3. Biologics
Rituximab has shown promise in treating IgG4-RD in multiple case series [52] and a prospective open-label study of 30 patients, which reported a 97% response rate with a significant reduction in the IgG4-RD Responder Index (IgG4-RD RI). The primary outcome, defined as a decrease in IgG4-RD RI >2, no disease flare before month 6, and being off glucocorticoids between months 2 and 6, was achieved in 77% of patients. Other investigators have reported similar results, supporting a significant role for B cell depletion in IgG4-RD [53]. In clinical practice, rituximab is often considered the first-line steroid-sparing agent after a relapse, especially in regions where it is readily available [54]. Rituximab can be a valuable tool in managing relapsing IgG4-RD and reducing steroid dependence.
Bortezomib, a proteasome inhibitor initially used for multiple myeloma treatment, has been reported to be effective in a patient with IgG4-RD refractory to steroids and surgery, leading to clinical improvement in orbital pseudotumor and pulmonary infiltration [55]. However, bortezomib was used in combination with cyclophosphamide, making it unclear if bortezomib was independently effective. Yamamoto et al. described the effectiveness of abatacept in a rituximab-refractory IgG4-RD patient, suggesting a potentially important role for T cells in IgG4-RD pathogenesis. This patient had complete CD19+ cell depletion yet still relapsed [56]. While the mechanism of abatacept’s benefit is unknown, it may involve modulation of follicular helper T cells and induction of indoleamine 2,3-dioxygenase secretion by dendritic cells and macrophages, which can inhibit T cell proliferation [57, 58].
Infliximab, a monoclonal antibody against tumor necrosis factor-alpha, has been proposed for treating corticosteroid-refractory IgG4-related orbital disease. Infliximab is known to be beneficial in severe orbital inflammation unresponsive to conventional immunosuppression and may have beneficial effects in IgG4-related orbital disease by interfering with IgG4 production in activated plasma cells and through Th2 lymphocyte modulation [59]. Biologic therapies like rituximab, abatacept, and infliximab represent promising alternatives for patients with refractory or relapsing IgG4-RD, but further research is needed to define their optimal roles in management algorithms.
4. Radiation
Radiation therapy has historically been used for orbital pseudotumors. With improved understanding of IgG4-RD and its responsiveness to glucocorticoids and B cell depletion, clinicians now try to avoid radiation therapy. However, radiation may still be beneficial in steroid-intolerant cases. A recent case series of IgG4-related ocular adnexal disease reported clinical improvement and steroid tapering with minimal complications using radiation [60]. Radiation therapy remains a consideration in specific scenarios, particularly for localized disease unresponsive to or unsuitable for medical therapies.
5. Stents
IgG4-RD is a fibroinflammatory condition, and delayed treatment can lead to significant fibrosis and irreversible organ damage that may not respond to glucocorticoids or immunosuppressants. Fibrotic tissue or masses can cause mechanical obstruction and organ dysfunction that may require mechanical relief. In obstructive jaundice secondary to IgG4-related cholangitis, endoscopic placement of metal or plastic stents can relieve biliary strictures and obstruction [61]. Similarly, in IgG4-RD retroperitoneal fibrosis with ureteral obstruction, ureteral stenting has shown benefit in case reports [62, 63]. These measures can provide temporary relief in acute presentations while medical treatments take effect. In some cases of end-stage disease, stenting may be the only viable option. Mechanical interventions like stenting can be critical adjuncts to medical management in addressing obstructive complications of IgG4-RD.
5. PROGNOSIS
The natural course of IgG4-RD is not fully defined due to limited long-term studies. Case reports and series suggest that some patients may experience transient improvement without treatment, but many experience disease recurrence in the same or different organs, and some develop chronic progressive disease [64]. It is crucial to assess for multi-organ involvement in patients presenting with single-organ disease because untreated involvement in certain organs can lead to irreversible damage. Without prompt treatment, complications such as cirrhosis and portal hypertension, retroperitoneal fibrosis, aortic aneurysm complications, biliary obstruction, and diabetes mellitus can cause significant morbidity and mortality. Further studies are needed to better define the long-term prognosis of IgG4-RD.
Recent studies suggest a potentially increased risk of malignancy in IgG4-RD patients compared to the general population. Yamamoto et al. and Huggett et al. reported malignancy rates of 10.4% and 11% in their IgG4-RD cohorts over 3 years, with an estimated risk approximately 3 times higher than in the general population [65]. A study by Asano et al. [66] reported an even higher rate of 22% over a mean follow-up of 6 years. The older age of IgG4-RD patients and frequent cross-sectional imaging for disease monitoring might contribute to bias in these studies. More detailed investigations are required to accurately assess the relationship between IgG4-RD and malignancy risk. Long-term follow-up and cancer surveillance may be warranted in IgG4-RD patients, particularly those with specific organ involvement or risk factors.
6. CONCLUSION
Tissue biopsy remains the gold standard for igg4 diagnosis, but robust clinicopathological correlation is essential for confirming IgG4-RD [Figure 2]. Increased clinician awareness of IgG4-RD will improve recognition and prompt diagnosis, which is crucial for preventing organ damage. The absence of a reliable biomarker has been a significant diagnostic challenge. Advances in understanding IgG4-RD pathogenesis, particularly the roles of plasmablasts and T-B cell interactions, offer promising avenues for developing new biomarkers and targeted therapies. The optimal steroid-sparing medications for IgG4-RD remain uncertain due to a lack of randomized controlled trials. However, with increased awareness and international collaborations, the field is progressing towards conducting clinical trials to evaluate maintenance therapies for IgG4-RD.
Figure 2. Summary of key elements in diagnosis of IgG4-RD.
*Gold Standard and still necessary for diagnosis
Expert Commentary
IgG4-RD often presents as tumor-like lesions, mimicking malignancies, infectious disorders, or inflammatory conditions like GPA (Wegener’s). Many patients undergo invasive procedures, such as resection or biopsy of the affected organ, to rule out other conditions. Unfortunately, after receiving reassurance that malignancy is excluded, some patients are discharged without a definitive igg4 diagnosis. Many of these patients later experience recurrence in other organs within months or years, requiring further extensive and costly investigations, or they present with organ dysfunction due to progressive fibrosis, leading to irreversible damage such as cirrhosis, renal failure, or vision loss.
IgG4-RD, while under-recognized, is not a rare disease. Estimating the true population burden of IgG4-RD, including incidence and prevalence, is challenging due to its recent recognition. Current epidemiological data is limited, mainly from Japanese studies focused on autoimmune pancreatitis. Improved igg4 diagnosis rates are hampered by insufficient awareness of this condition. Definitive diagnosis typically requires biopsy, expert pathological interpretation considering IgG4-RD, and thorough clinicopathological correlation with patient history, radiology, and laboratory findings. Establishing these diagnostic pathways takes time and requires increased awareness. Conversely, identifying a reliable biomarker would significantly facilitate diagnosis through suggestive laboratory testing. Since its initial recognition, and even currently in Japan, elevated serum IgG4 concentrations and increased IgG4-positive plasma cells in tissue have been considered diagnostic hallmarks. However, the reported frequency of elevated serum IgG4 in published series varies widely, from 44% to 100%. Recently, Western investigators have increasingly questioned the diagnostic utility of serum IgG4 due to assay inaccuracies and variability in reported values.
Nonetheless, elevated serum IgG4 remains highly suggestive of IgG4-RD, especially in patients with characteristic organ involvement, such as orbital tumor-like lesions. Elevation greater than twofold the upper limit of normal (normal: 140 mg/dL) has a high specificity (99%) for IgG4-RD and is rarely seen in other conditions. Furthermore, in many patients, serum IgG4 concentrations correlate approximately with disease activity. In patients with multi-organ involvement, serum IgG4 elevation can be dramatic, occasionally reaching 30 to 40 times the upper limit of normal.
No single test or clinical feature definitively diagnoses IgG4-RD. The lack of a reliable biomarker complicates igg4 diagnosis even when clinicians are aware of the condition. Diagnostic delays in IgG4-RD can lead to serious complications, including cirrhosis, pancreatic failure, thoracic or abdominal aortic aneurysms, advanced renal dysfunction, and others. The limitations of serum IgG4 assays in accurately identifying potential IgG4-RD patients can also lead to increased morbidity and costs from unnecessary diagnostic procedures. Therefore, improved understanding of current diagnostic tests and the importance of clinicopathological correlation, as summarized in this review, can enhance igg4 diagnosis and disease activity monitoring.
A defining feature of IgG4-RD is its marked responsiveness to steroid treatment. This response is often dramatic and is included in some diagnostic criteria for IgG4-RD. While glucocorticoids induce complete remission in many patients, there is a critical need for effective steroid-sparing maintenance therapies. Glucocorticoid side effects are well-known and particularly concerning in this older patient population, many of whom have pre-existing diabetes due to pancreatic insufficiency. This review has summarized current treatment strategies commonly used by IgG4-RD experts.
5-year view
IgG4-RD has gained official recognition within the medical community in the past decade. Significant progress has been made in its clinical description and treatment responsiveness. Ongoing research into disease mechanisms will guide future advancements in identifying reliable biomarkers for igg4 diagnosis and improving management strategies over the next five years.
KEY ISSUES.
- The true US prevalence of IgG4-RD is unknown, but it is thought to affect males more than females and have an age of onset between 50–70 years old.
- Organs can be affected at different times (metachronously), often leading to clinical and diagnostic confusion.
- Elevated serum IgG4 level is neither necessary nor sufficient for igg4 diagnosis. In cases with low IgG4 serum levels but high clinical suspicion, the prozone effect should be considered.
- Measuring circulating plasmablasts or the IgG4/IgG RNA ratio may be superior biomarkers for active IgG4-RD compared to serum IgG4 levels.
- Clinicohistopathological findings remain the diagnostic gold standard for this disorder.
- Glucocorticoids are the best initial therapy, although disease relapse is common.
- Rituximab has demonstrated benefit as a steroid-sparing treatment.
- Other steroid-sparing agents, such as methotrexate, azathioprine, and mycophenolate mofetil, are used for maintenance therapy, but their efficacy is not well-established.
- Surgical resection and stent placement can be used when medical treatment is insufficient.
Acknowledgments
Funding
This manuscript was funded by the National Institutes of Health, National Center for Advancing Translational Sciences (KL2TR000455 and UL1TR000454)
Footnotes
Declaration of interest
The authors declare that they have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Contributor Information
Mary Abraham, Emory University School of Medicine, 1365 Clifton Rd, 3rd floor, Atlanta, GA 30322.
Arezou Khosroshahi, Assistant Professor of Medicine, Emory University School of Medicine, 1365 Clifton Rd, 3rd floor, Atlanta, GA 30322.
References
*= of interest; **= of considerable interest