Juvenile myelomonocytic leukemia (JMML) diagnosis relies heavily on understanding its unique molecular underpinnings. This rare childhood blood cancer is strongly associated with specific genetic mutations, primarily within the RAS pathway. Identifying these molecular features is crucial for accurate diagnosis, prognosis, and potentially targeted therapeutic strategies for JMML.
The Central Role of RAS Pathway Mutations in JMML Diagnosis
The genomic landscape of JMML is notably defined by mutations in genes belonging to the RAS pathway. These genes – NF1, NRAS, KRAS, PTPN11, and CBL – are central to cell growth and signaling. Diagnostic investigations into suspected JMML cases frequently involve screening for variants in these genes.
Among these, PTPN11 stands out as the most frequently mutated gene in JMML. Studies analyzing a large cohort of JMML patients have revealed that PTPN11 mutations are present in approximately 51% of cases. These mutations can be either germline (inherited) or somatic (acquired). Specifically, around 19% are germline and 32% are somatic, highlighting the significant role of PTPN11 in JMML development.
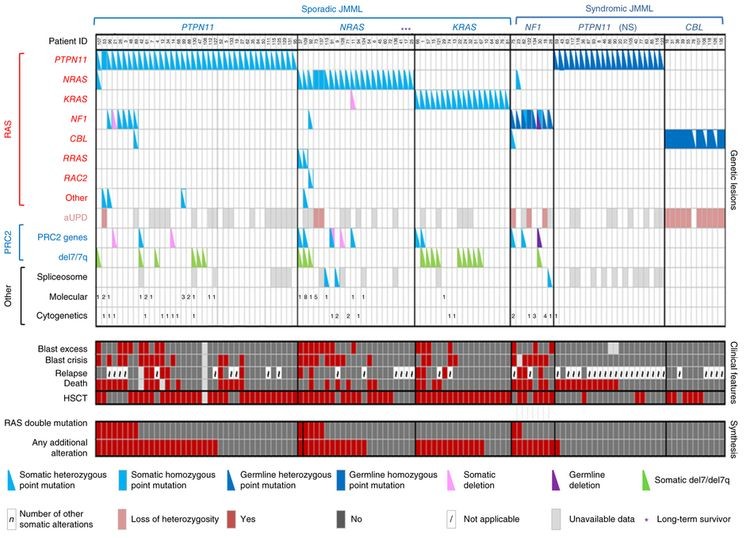 Alteration profiles in individual JMML cases for jmml diagnosis, highlighting RAS pathway mutations and their frequencies.
Alteration profiles in individual JMML cases for jmml diagnosis, highlighting RAS pathway mutations and their frequencies.
Further molecular diagnosis in JMML reveals that NRAS and KRAS mutations are also significant, found in 19% and 15% of cases respectively. Mutations in NF1 and CBL are observed in a smaller proportion, accounting for 8% and 11% of cases. It’s important to note that while these mutations are usually mutually exclusive, a small percentage of JMML cases (4% to 17%) can harbor mutations in two or more of these RAS pathway genes. This co-occurrence is clinically relevant as it has been linked to a less favorable prognosis, emphasizing the importance of comprehensive molecular profiling for accurate Jmml Diagnosis and risk stratification.
Secondary Genomic Alterations and JMML Prognosis
While RAS pathway mutations are the hallmark of JMML, the molecular picture can be more complex. JMML cells often exhibit a low overall mutation burden, but additional genetic alterations beyond the primary RAS pathway genes are frequently observed. These secondary mutations provide further insights into disease biology and prognosis in JMML diagnosis.
Mutations affecting genes of the Polycomb Repressive Complex 2 (PRC2), such as ASXL1, are found in a subset of JMML cases (7%–8%). Similarly, genes more commonly associated with adult myeloproliferative neoplasms, like SETBP1, are also mutated in a small percentage of JMML patients (6%–9%). JAK3 mutations are another category of secondary alterations detected in 4%–12% of JMML cases. Notably, patients with germline PTPN11 or CBL mutations tend to have fewer of these additional mutations.
The presence of these secondary genomic alterations alongside the primary RAS pathway mutations is clinically significant. They are often associated with a poorer prognosis, underscoring the importance of considering the full spectrum of molecular features in JMML diagnosis to accurately predict disease course and guide management strategies.
Rare Receptor Tyrosine Kinase Fusions in JMML Diagnosis
In a subset of JMML cases, particularly those lacking the typical RAS pathway mutations, alternative molecular drivers have been identified. Approximately 11% of JMML patients do not present with canonical RAS pathway mutations. Further investigation in these cases has revealed the presence of in-frame fusions involving receptor tyrosine kinases (RTKs).
Specifically, DCTN1::ALK, RANBP2::ALK, and TBL1XR1::ROS1 gene fusions have been identified in these RAS pathway-negative JMML cases. Intriguingly, these patients often share other clinical characteristics, such as monosomy 7 and older age at diagnosis (56 months or older). The identification of these RTK fusions is not only diagnostically relevant but also therapeutically actionable. For instance, one patient with an ALK gene fusion achieved complete molecular remission after treatment with crizotinib, an ALK inhibitor, combined with conventional chemotherapy, and subsequently underwent successful allogeneic bone marrow transplantation. This highlights the potential of precision medicine approaches in JMML diagnosis and treatment, particularly in molecularly defined subgroups.
Conclusion: Integrating Molecular Features into JMML Diagnosis
In conclusion, a comprehensive molecular understanding is integral to accurate JMML diagnosis. The presence and type of RAS pathway mutations, along with the identification of secondary genomic alterations and rare RTK fusions, provide critical diagnostic and prognostic information. As our understanding of the JMML molecular landscape expands, so too does our ability to refine diagnostic approaches, predict disease outcomes, and develop more targeted and effective therapies for this challenging pediatric leukemia.
