Congenital Long QT Syndrome (LQTS) is a heart condition characterized by an extended QT interval on an electrocardiogram (ECG). This elongation occurs in the absence of structural heart abnormalities or external factors like medications. LQTS was first identified in the 1950s, initially linked to deafness in the Jervell and Lange-Nielsen syndrome. Later, patients with similar ECG changes but without deafness (Romano-Ward syndrome) were recognized in the early 1960s. While initially separated, these conditions are now broadly termed LQTS. Genetic research since the mid-1990s has further refined our understanding, allowing for sub-classification based on specific genetic defects. The history of LQTS, spanning over six decades, is comprehensively reviewed by Dr. Schwartz.
Methods for Long QT Diagnosis
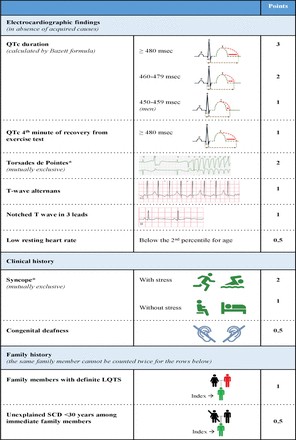
Diagnosing LQTS involves analyzing the heart rate-corrected QT interval (QTc) alongside other ECG parameters and patient history, including symptoms and family history. These elements combine to form the LQTS probability, or Schwartz score. A score of 3.5 points or higher indicates a high likelihood of LQTS (figures 1 and 2). Genetic information, while not part of the Schwartz score, is crucial; identifying a pathogenic variant is independently diagnostic for LQTS. It’s critical to understand that QTc values in individuals with pathogenic variants and healthy individuals can overlap. This overlap means a single QTc measurement cannot definitively distinguish all LQTS ECGs from non-LQTS ECGs. Furthermore, QT interval measurement methods and heart rate correction formulas vary, leading to different cut-off values. While U-waves can be abnormal in LQTS, they should not be included in QT interval measurements. To aid in diagnosis, an online calculator based on a large dataset of genotyped LQTS patients and their families is available (https://www.qtcalculator.org). This tool provides LQTS likelihood based on the calculated QTc. Recent advancements have introduced additional tools, including artificial intelligence, to enhance the reliability of LQTS diagnosis via ECG.
Figure 1: Diagnostic criteria for Long QT syndrome (LQTS), also known as the Schwartz score, which is used to evaluate the likelihood of LQTS. A score of ≥3.5 indicates a high probability of LQTS.
Figure 2: Diagnostic flow chart for LQTS, illustrating the diagnostic process following international guidelines, incorporating ECG analysis and risk assessment to determine the likelihood of LQTS.
Genetic Testing and Phenotype Correlations in LQTS Diagnosis
Over the past 25 years, 17 genes have been linked to LQTS. However, rigorous analysis by the Clinical Genome Resource (ClinGen) has reclassified several of these, identifying seven genes with definitive or strong causal evidence. These confirmed genes are involved in cardiac repolarization, encoding ion channels, their regulatory subunits, or proteins that modulate ion channel function. Positive genetic testing, achievable in up to 75% of phenotype-positive individuals, is diagnostically significant and informs risk assessment and treatment strategies.
Specific genotype-phenotype correlations are well-documented for the three most common LQTS subtypes: types 1, 2, and 3 (figure 3). LQT1 and LQT2 arise from loss-of-function variants in potassium channel genes KCNQ1 and KCNH2, respectively. These genes encode for potassium currents IKs and IKr, and reduced current amplitude leads to QT interval prolongation (figure 4A–D). LQT3 results from gain-of-function variants in SCN5A, the gene for the cardiac sodium current (INa). This gain of function increases the late sodium current during the plateau phase of the action potential, also prolonging repolarization.
Figure 3: Genotype-phenotype relationship in LQTS types 1, 2, and 3. This chart summarizes the distinct clinical features, triggers for arrhythmias, and therapeutic responses associated with each major genetic subtype of LQTS.
Figure 4: Pathophysiological mechanisms in LQTS and related arrhythmias. This figure explains how genetic defects affect cardiac ion currents, prolong action potentials, and predispose individuals to dangerous arrhythmias like Torsades de Pointes.
Arrhythmia onset age varies by subtype; LQT1 patients, particularly males, are at risk at a younger age. LQT2 and LQT3 patients often experience initial symptoms around puberty, with females being more susceptible in these subtypes (figure 3). ST-T segment morphology is also subtype-specific, allowing for genotype prediction based on ECG features. Each genotype has distinct arrhythmia triggers and ECG characteristics at arrhythmia onset (figure 3). LQTS arrhythmias (figure 4F) originate from prolonged ventricular action potentials, leading to early afterdepolarizations and triggered beats that can degenerate into Torsades de Pointes and ventricular fibrillation. Adrenergic triggers like exercise and swimming are common in LQT1, while sudden arousal (auditory stimuli) is typical in LQT2 (figure 3). LQT3 events often occur at rest. These genotype-specific traits influence treatment responses. LQT1 patients show high responsiveness to β-blockers, which remain first-line therapy for all subtypes. LQT3 patients are particularly sensitive to late sodium channel blockers. Given these genotype-specific implications for prognosis and treatment (figure 3), genetic testing is essential for LQTS diagnosis and management.
Clinical Management, Treatment Strategies, and Diagnostic Relevance
Beta-blocker therapy is the cornerstone of LQTS management (figure 5). Non-selective beta-blockers like nadolol and propranolol are often preferred. While propranolol’s efficacy has been debated, its perceived lower effectiveness may be skewed by its use in severe neonatal LQTS cases. Metoprolol and atenolol are considered less effective, especially in symptomatic patients. Beta-blockers prevent early afterdepolarizations by blocking adrenergic-driven calcium current increases. Propranolol also offers some QTc reduction by blocking late sodium current. Treatment is clearly indicated for symptomatic patients. However, the necessity of treatment for asymptomatic individuals, increasingly identified through cardiogenetic screening, is less clear.
Figure 5: Therapeutic flow chart for LQTS, outlining treatment decisions according to international guidelines, from pharmacological interventions to device therapy, based on patient risk and response to initial treatments.
Additional pharmacological options include late sodium current blockers (mexiletine, flecainide, ranolazine). LQT3 patients are particularly responsive, but mexiletine has also shown QTc reduction in LQT2. Sodium channel blockers can, however, induce a Brugada ECG pattern, requiring careful evaluation, especially in SCN5A variant carriers with mixed function characteristics. Potassium supplementation is beneficial in LQT2 due to the potassium-dependent nature of IKr conductance. Maintaining adequate extracellular potassium levels is important across LQTS subtypes.
New therapies are emerging, often targeting specific patient subgroups. Lumacaftor has shown promise in shortening QTc in patients with KCNH2 trafficking defects by improving intracellular protein transport.
Avoiding drugs that reduce repolarization reserve and managing electrolyte imbalances, particularly hypokalemia, are critical in LQTS management (figure 4E). Drug-induced QTc prolongation can be exacerbated by LQTS-causing mutations and hypokalemia (figure 6A,B). CredibleMeds (www.QTdrugs.org) provides resources on QT-prolonging drugs, though its limitations should be recognized.
Figure 6: Effects of genetic and environmental factors in LQTS. This diagram illustrates how genetic mutations and environmental factors like drugs or electrolyte imbalances can synergistically prolong the QT interval, and how protective genetic factors may mitigate these effects.
Left cardiac sympathetic denervation (LCSD) is considered when beta-blockers are insufficient. It is recommended for high-risk LQTS patients when ICD therapy is contraindicated or refused, or when beta-blockers are ineffective, not tolerated, or contraindicated. LCSD can also be useful for breakthrough events despite beta-blocker/ICD therapy. It is particularly effective in LQT1 but can benefit LQT2, LQT3, and gene-elusive LQTS patients.
Implantable cardioverter-defibrillators (ICDs) are the final therapy level for patients with breakthrough events or high arrhythmia risk. ICDs are class 1 recommended for cardiac arrest survivors (with beta-blockers; figure 5), possibly except for undiagnosed LQT1 patients at the time of arrest. ICDs are effective in preventing sudden cardiac death, but complications occur at a rate of 7.0% per year, with inappropriate shocks in 3% of patients annually.
Pacemaker therapy is less studied in LQTS but may benefit LQT2 patients with pause-dependent arrhythmias. Pause-dependent pacing algorithms have shown success in individual cases. In LQT3, where QTc prolongation is more pronounced at lower heart rates, pacing at higher rates may be beneficial. This has been demonstrated in a large LQT3 family. Atrial pacing is also effective in severe Jervell Lange-Nielsen syndrome, even in young children. Temporary high-rate pacing can also be effective during arrhythmic storms.
Future Directions in Long QT Diagnosis and Research
LQTS knowledge has advanced significantly, transforming it from an ECG curiosity with high sudden death risk to a genotype-specific disease with refined risk stratification and treatment, leading to reduced mortality. However, treatment optimization is needed, particularly for treatment-resistant severe LQTS, often seen in neonates with specific mutations or double mutations. New drugs are needed for this subgroup. Conversely, improved risk stratification is crucial for the growing population of asymptomatic individuals identified through cardiogenetic programs, many with normal or borderline QTc, to determine appropriate management.
Furthermore, beta-blocker adherence is suboptimal, reported at only 50% in some studies. Side effects, illness perception, and medication beliefs contribute to non-adherence, impacting arrhythmia risk. Non-compliance and QT-prolonging drug use are major factors in beta-blocker failures in LQT1. LCSD as standalone therapy may be an option for patients with severe beta-blocker side effects.
Despite genetic screening advances, pathogenic variants are identified in only 75%-80% of phenotype-positive LQTS cases, and even lower in recent clinic data due to broader testing criteria. Ongoing research is vital to unravel the genetic basis of gene-elusive cases and to understand additional factors influencing arrhythmia risk. Comorbidities like hypertension may worsen LQTS phenotypes. Polygenic architecture may play a role in genotype-negative patients, and protective and deleterious alleles may modify LQTS severity (figure 6B,C). Identifying new genes remains crucial for understanding gene-elusive LQTS.
In summary, congenital LQTS is an inherited condition characterized by prolonged QTc and associated with malignant arrhythmias in young individuals. It results from complex polygenic influences on cardiac repolarizing currents, interacting with factors like sex, age, comorbidities, and triggers. Therapy relies on beta-blockers and lifestyle modifications, with advancing patient-specific therapies under development. Enhanced understanding of LQTS complexity is paving the way for improved patient and family care through better Long Qt Diagnosis strategies and personalized treatments.