Introduction
The 2016 revision of the World Health Organization (WHO) classification system for hematopoietic and lymphoid tissue tumors brought significant updates to the diagnostic criteria for myeloproliferative neoplasms (MPNs). Published in September 2017, this revised classification refined the categories within MPNs, which now include chronic myeloid leukemia, chronic neutrophilic leukemia, polycythemia vera (PV), primary myelofibrosis (PMF), essential thrombocythemia (ET), chronic eosinophilic leukemia-not otherwise specified, and MPN-unclassifiable. Notably, mastocytosis was reclassified and is no longer under the MPN umbrella. This article focuses specifically on the Myelofibrosis Diagnosis Criteria, alongside those for PV and ET, particularly in the context of JAK2, CALR, and MPL mutations. The 2016 revisions aimed to improve the differentiation between conditions like masked PV and JAK2-mutated ET, and between the early (prefibrotic) and advanced (overtly fibrotic) stages of PMF. This discussion will provide a practical overview of these updated myelofibrosis diagnosis criteria, explore the reasoning behind the changes, examine the role of mutation analysis, address existing controversies, and consider the practical challenges and future directions for clinical pathologists.
2016 WHO MPN Subcategories and Overview of Diagnostic Criteria
In the 2016 WHO classification, morphology remains the cornerstone for diagnosing hematopoietic and lymphoid tissue tumors. However, genetic mutation screening is increasingly vital, serving to confirm morphologic diagnoses and, in some cases, guiding the diagnostic process itself. Myeloid neoplasms continue to be categorized into acute and chronic forms based primarily on blast percentages in peripheral blood and bone marrow. Chronic myeloid neoplasms are further divided into myelodysplastic syndromes (MDS), MPNs, MDS/MPN overlap syndromes, and myeloid/lymphoid neoplasms with eosinophilia and specific genetic rearrangements. MPNs are generally distinguished from MDS and MDS/MPN by the absence of significant dysplasia, such as dyserythropoiesis, dysgranulopoiesis, and monocytosis.
Within the MPN category, the 2016 WHO classification emphasizes the JAK2, CALR, and MPL mutation-related MPNs: PV, ET, and PMF. These entities, along with chronic myeloid leukemia (CML), chronic neutrophilic leukemia (CNL), chronic eosinophilic leukemia not otherwise specified (CEL-NOS), and MPN-unclassifiable (MPN-U), constitute the MPN spectrum. Our primary focus here is on the diagnostic criteria for myelofibrosis, PV, and ET, particularly in relation to JAK2, CALR, and MPL mutations.
CML is defined by the consistent presence of the BCR-ABL1 mutation. While minor BCR-ABL1 clones can sometimes be found in other myeloid neoplasms, including JAK2/CALR/MPL-mutated MPNs, these do not typically override the primary morphologic diagnosis. Conversely, JAK2 mutations can occasionally be detected in CML patients, particularly after successful imatinib treatment.
CNL is characterized by the clonal proliferation of mature neutrophils, frequently associated with activating mutations in the CSF3R gene, most commonly T618I. These CSF3R mutations appear to be specific to WHO-defined CNL. Diagnosing CNL requires excluding other causes of neutrophilia, such as infections, inflammatory conditions, metastatic cancer, and plasma cell neoplasms. Mature neutrophilia can also occur in other myeloid malignancies like atypical CML and chronic myelomonocytic leukemia. The 2016 WHO diagnostic criteria for CNL are designed to differentiate it from both secondary neutrophilia and clonal neutrophilia associated with other myeloid malignancies. Key criteria include leukocytosis (≥25 × 109/L) with ≥80% segmented/band neutrophils, and absence of eosinophilia and basophilia. The presence of a membrane proximal CSF3R mutation in neutrophilic granulocytosis is often sufficient for CNL diagnosis, regardless of leukocytosis levels.
CEL-NOS is diagnosed in the presence of clonal eosinophilia, defined as ≥1.5 × 109/L absolute eosinophil count in peripheral blood, along with either myeloblast excess (>2% in peripheral blood or 5–19% in bone marrow) or a clonal cytogenetic abnormality. Common cytogenetic abnormalities include trisomy 8, t(10;11)(p14;q21), and t(7;12)(q11;p11). Next-generation sequencing suggests some cases of “hypereosinophilic syndrome” may be reclassified as CEL-NOS. Unlike PDGFRA/B-rearranged myeloid/lymphoid neoplasms with eosinophilia, CEL-NOS does not respond to imatinib therapy.
MPN-U encompasses MPN-like neoplasms that cannot be definitively classified into the other six MPN subcategories. Patients with MPN-U may present with unexplained thrombosis, particularly splanchnic vein thrombosis, often with normal blood counts.
Practical Diagnostic Criteria for PV, ET, and PMF (Including Prefibrotic PMF)
The WHO emphasizes the integrated approach using clinical, morphologic, and molecular genetic features for defining MPN entities. Tables 1 and 2 summarize the 2016 WHO diagnostic criteria for PV, ET, and PMF, including prefibrotic/early PMF (pre-PMF). These criteria have been validated by numerous clinical-pathological studies, highlighting the significance of morphological features. However, debates persist regarding the strictness and application of these guidelines. Some have argued that bone marrow morphology alone may not reliably distinguish ET, PV, and PMF due to their potential to transform into one another. The notion that JAK2-mutated ET closely resembles PV in hematological presentation and clinical manifestations has also been discussed, although this perspective often arises from studies not strictly adhering to WHO criteria.
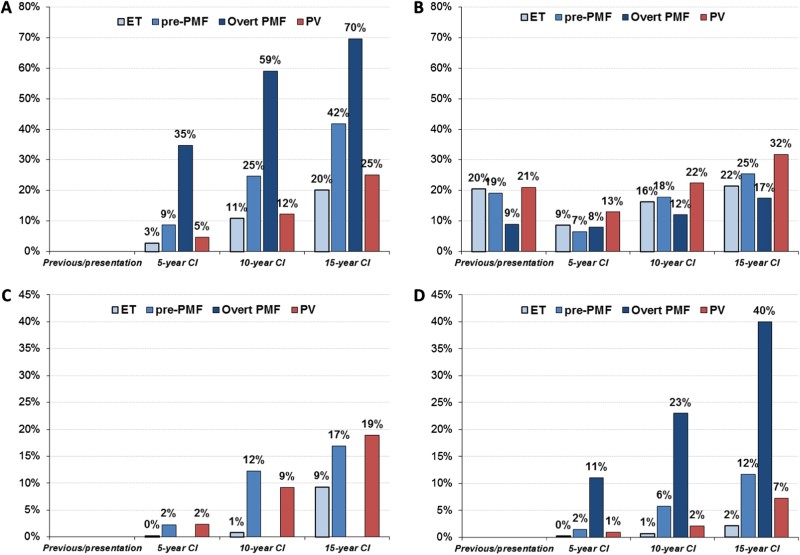
Studies strictly applying WHO criteria show that transformation of ET to PV is rare, occurring in approximately 1% of wild-type ET and up to 5% of JAK2-mutated ET after two decades of follow-up. Differentiating ET from pre-PMF is crucial, supported by distinct bone marrow morphology and differing clinical behaviors (Fig. 1a-d). ET generally exhibits a more benign course regarding survival, myelofibrosis progression, and blast phase transformation. However, major thrombosis risk in ET is comparable to pre-PMF and lower than PV. Conversely, pre-PMF demonstrates a clear progression pattern distinct from ET, with higher rates of evolution to overt PMF, blast crisis, and mortality (Fig. 1a, c, d), along with an increased bleeding tendency. PV, including masked PV (mPV), shows a trend towards more frequent thrombotic events and a higher incidence of progression to myelofibrosis. Overt PMF has the highest mortality and blast crisis transformation rates, but a lower cumulative incidence of thrombotic complications (Fig. 1a, b, d).
Table 1. 2016 World Health Organization Diagnostic Criteria for Polycythemia Vera and Essential Thrombocythemia
| Polycythemia vera (PV) a | Essential thrombocythemia (ET) b |
|---|---|
| Major criteria | |
| 1 | Hemoglobin > 16.5 g/dL (men) Hemoglobin > 16.0 g/dL (women) or Hematocrit > 49% (men) Hematocrit > 48% (women) or increased red cell mass (RCM)c |
| 2 | BM biopsy showing hypercellularity for age with trilineage growth (panmyelosis) including prominent erythroid, granulocytic and megakaryocytic proliferation with pleomorphic, mature megakaryocytes (differences in size) |
| 3 | Presence of JAK2 or JAK2 exon 12 mutation |
| 4 | Presence of JAK2, CALR or MPL mutation |
| Minor criteria | |
| 1 | Subnormal serum erythropoietin level |
Table adapted from Barbui T et al. Blood Cancer J 2015; 5:e337 and Arber et al. Blood 2016;127:2391–2405
BM, bone marrow; CML, chronic myeloid leukemia; MDS, myelodysplastic syndrome
aPV diagnosis requires meeting either all three major criteria or the first two major criteria and one minor criterion
bET diagnosis requires meeting all four major criteria or first three major criteria and one minor criterion
cMore than 25% above mean normal predicted value
dGrading of BM fibers
Criterion number 2 (BM biopsy) may not be required in cases with sustained absolute erythrocytosis: hemoglobin levels. 18.5 g/dL in men (hematocrit, 55.5%) or 16.5 g/dL in women (hematocrit, 49.5%) if major criterion 3 and the minor criterion are present. However, initial myelofibrosis (present in up to 20% of patients) can only be detected by performing a BM biopsy; this finding may predict a more rapid progression to overt myelofibrosis (post-PV MF)
Table 2. 2016 World Health Organization Diagnostic Criteria for Primary Myelofibrosis
| Primary myelofibrosis (PMF) a |
|---|
| Prefibrotic/early PMF (pre-PMF) |
| Major criteria |
| 1 |
| 2 |
| 3 |
| Minor criteria |
| 1 |
| • Anemia not attributed to a comorbid condition |
| • Leukocytosis ≥ 11 × 109/L |
| • Palpable splenomegaly |
| • LDH level above the upper limit of the institutional reference range |
| • Leukoerythroblastosis |
Table adapted from Barbui T et al. Blood Cancer J. 2015; 5:e337 and Arber et al. Blood 2016;127:2391–2405
BM, bone marrow; CML, chronic myeloid leukemia; MDS, myelodysplastic syndrome; LDH, serum lactate dehydrogenase
aDiagnosis of prefibrotic/early PMF requires all three major criteria and at least one minor criterion. Diagnosis of overt PMF requires meeting all three major criteria and at least one minor criterion
bSmall-to-large megakaryocytes with aberrant nuclear/cytoplasmic ratio and hyperchromatic and irregularly folded nuclei and dense clustering
cIn cases with grade 1 reticulin fibrosis, the megakaryocyte changes must be accompanied by increased BM cellularity, granulocytic proliferation, and often decreased erythropoiesis (that is, pre-PMF)
dIn the absence of any of the three major clonal mutations, the search for the most frequent accompanying mutations (ASXL1, EZH2, TET2, IDH1/IDH2, SRSF2, SF3B1) are of help in determining the clonal nature of the disease
eMinor (grade 1) reticulin fibrosis secondary to infection, autoimmune disorder or other chronic inflammatory conditions, hairy cell leukemia or other lymphoid neoplasm, metastatic malignancy, or toxic (chronic) myelopathies
fBM fibrosis secondary to infection, autoimmune disorder, or other chronic inflammatory conditions, hairy cell leukemia, or other lymphoid neoplasm, metastatic malignancy or toxic (chronic) myelopathies
Fig. 1.
 Mortality, Thrombosis, Myelofibrosis, and Blast Transformation in MPNs
Mortality, Thrombosis, Myelofibrosis, and Blast Transformation in MPNs
Mortality a, major arterial and venous thrombotic complications b, myelofibrosis progression c, and Blast transformation d in ET, Pre-PMF, overt PMF, and PV cohorts. Prevalence of previous events and cumulative incidence (CI) during follow-up calculated at 5, 10, and 15 years from diagnosis. For PMF, two different data sets were considered: n = 707 for panel a, b and n = 383 for panel d and regarding PV for all panels
Rationale for the 2016 Diagnostic Criteria Changes
The 2016 WHO revisions built upon the 2008 guidelines, incorporating insights from clinicopathological and molecular genetic studies. These updates addressed critical questions and borderline cases identified in reviews of MPNs. Concerns were raised about the diagnostic specificity of bone marrow morphology for MPN differentiation, aside from ruling out myelodysplasia. It is important to understand that MPN transformation does not inherently invalidate the initial diagnosis but rather reflects disease evolution. For instance, masked PV (mPV) may initially resemble ET before progressing to overt PV, and pre-PMF can present with an ET-like phenotype before evolving into overt PMF. These apparent “instabilities” in MPN subtyping are significantly influenced by the accuracy of the initial diagnosis.
The Role and Limitations of Mutation Screening in Myelofibrosis Diagnosis
The 2008 WHO classification was significantly influenced by the discovery of JAK2 mutations (V617F in exon 14 and insertions/deletions in exon 12) and MPL mutations (primarily W515 in exon 10). These mutations became major diagnostic criteria. JAK2V617F is the most common MPN mutation, found in ~95% of PV, and ~60% of ET and PMF cases. JAK2 exon 12 mutations occur in ~3-5% of JAK2V617F-negative PV patients. MPL mutations are found in approximately 4% of ET and 8% of PMF.
In 2013, CALR mutations, located in exon 9, were identified in JAK2/MPL-unmutated ET and PMF. The two most prevalent CALR mutation types are type 1 (52-bp deletion) and type 2 (5-bp insertion), accounting for over 80% of all CALR variants. Type 2 mutations are more common in ET, while type 1 predominates in PMF. These three driver mutations (JAK2V617F, JAK2 exon 12, CALR, and MPL) are major criteria for PV, ET, and PMF in the 2016 WHO classification.
Modern myelofibrosis diagnosis, as well as diagnosis of other MPNs, relies heavily on mutation status. However, when mutation testing is unavailable or negative, minor criteria in the WHO classification become crucial for supporting a diagnosis. Some PV patients lacking JAK2 mutations may have other JAK2 mutations or mutations in genes like SH2B3/LNK. Up to 20% of ET and 10–15% of PMF patients are “triple-negative” (TN) for JAK2, CALR, and MPL mutations. While some TN cases may harbor non-canonical MPL and JAK2 mutations, these account for a minority. For triple-negative PMF, the 2016 WHO classification suggests investigating “most frequent” non-driver mutations like ASXL1, EZH2, TET2, IDH1/IDH2, SRSF2, SF3B1, which can indicate clonality. These mutations lack disease specificity and mutual exclusivity but are found in about 50% of PMF cases. Chromosomal abnormalities can also serve as markers of clonality.
While these additional mutations are not currently diagnostic criteria for PV or ET lacking driver mutations, studies show that around 50% of these patients carry such mutations. The interpretation of these genetic variants is complicated by clonal hematopoiesis of indeterminate potential (CHIP), reflecting age-related mutation accumulation. However, in the context of MPN hematologic abnormalities, finding these mutations supports the presence of pathologic clonal hematopoiesis.
Controversies Regarding PV and Prefibrotic PMF Diagnosis
Lowering the hemoglobin (Hb) threshold for PV diagnosis in the 2016 WHO revision (>16.5 g/dL for men and >16.0 g/dL for women) has raised concerns. Critics argue this could lead to excessive investigations in healthy individuals. Population studies analyzing complete blood counts (CBCs) indicated that a significant percentage of males (4.1-5.6%) and females (0.22-0.35%) in Canadian and Brazilian populations would meet these Hb thresholds, potentially dramatically increasing PV incidence. However, these studies were based on routine CBCs without JAK2 mutation status and were not derived from clinicopathological databases aligned with WHO Hb thresholds.
Conversely, studies on masked PV (mPV) show that many cases meeting 2016 WHO criteria were previously missed. A study of 118 mPV patients, 72% had a history of thrombosis, with thrombocytosis being the most common finding (64%). Thrombocytosis complicates the differentiation between mPV and ET, a challenge recognized before the 2016 revision. Misdiagnosing mPV as ET can lead to the inappropriate omission of phlebotomies, a crucial treatment for PV to maintain a therapeutic hematocrit target. Recognizing early PV stages is a significant advancement, preventing underdiagnosis and fatal thrombotic events by enabling timely treatment initiation.
The concept of prefibrotic PMF (pre-PMF) also faces challenges in widespread acceptance. Despite initial descriptions in the late 1990s and subsequent validation, pre-PMF is not universally recognized, including in updated British guidelines. The 2008/2016 WHO classifications define pre-PMF by absent or minor reticulin fibrosis (grade 0-1), while overt PMF shows grade 2-3 fibrosis including collagen. Difficulties in accepting pre-PMF as a distinct entity may stem from its diagnosis relying heavily on morphology and variable clinical presentations. Pre-PMF patients may present with an ET-like phenotype or a more PMF-like phenotype without thrombocytosis. Molecular markers for pre-PMF differ from ET but have limited discriminant power.
Recent research confirms that pre-PMF clinical presentation differs from ET, impacting treatment decisions and outcomes. Differentiating pre-PMF and ET is complex but crucial, affecting laboratory data, hemorrhage risk, thrombosis, and disease progression. Initial laboratory data distinguishes pre-PMF from WHO-defined ET. Minor criteria for pre-PMF (anemia, leukocytosis, elevated LDH, splenomegaly) are highly prevalent in pre-PMF (91%) compared to ET (48%). Higher circulating CD34+ cell counts and in vitro stem cell growth in pre-PMF further suggest distinct biological behavior. The unfavorable prognostic impact of JAK2V617F in pre-PMF versus the more benign course of CALR-mutated pre-PMF (not seen in ET) also supports this distinction.
Blood tests can have predictive power in ET-like presentations. Algorithms using Hb, WBC count, and LDH level have shown ~50% sensitivity and specificity in distinguishing pre-PMF. A logistic regression model improved this to ~75%. While bone marrow biopsy remains essential, laboratory parameters can aid in suspecting pre-PMF in presumptive ET cases. Survival, blast transformation, and progression to overt myelofibrosis rates are significantly worse in pre-PMF than WHO-confirmed ET. Risk scoring systems for overt PMF may not be applicable to pre-PMF due to differing clinical phenotypes. Different clinical pictures and outcomes necessitate different treatment approaches for pre-PMF and ET, highlighted by differing outcomes in studies comparing hydroxyurea and anagrelide in ET patients defined by older PVSG criteria (including many pre-PMF cases) versus WHO-classified ET.
Ongoing Controversies Regarding Bone Marrow Morphology in MPN Diagnosis
Concerns exist that bone marrow trephine biopsy in JAK2-mutated patients with marked erythrocytosis may be unnecessary and hazardous. It’s also argued that morphology lacks sufficient diagnostic specificity for differentiating PV from other MPNs and provides limited prognostic information. However, concerns about biopsy complications appear unsubstantiated.
A blinded review study showed high reproducibility of characteristic PV bone marrow features, with ~93% interobserver agreement. This study included mPV, overt PV, JAK2-mutated ET, and JAK2-mutated patients not meeting 2008 WHO Hb thresholds but confirmed as PV by red cell mass. Bone marrow biopsy also provides prognostic information, particularly regarding bone marrow fibrosis. While variable fibrosis incidence and severity have been reported, older studies often included advanced post-PV myelofibrosis stages. The clinical impact and prognostic relevance of reticulin fibrosis at PV diagnosis outset has been demonstrated in over 500 WHO-defined PV patients. Grade 1 reticulin fibrosis was found in 14% of patients, with higher grades being rare. While clinical and laboratory characteristics were similar with or without fibrosis, splenomegaly was more prevalent in fibrotic cases, and these patients more frequently progressed to post-PV myelofibrosis. These findings were recently validated, emphasizing the association between bone marrow reticulin fibrosis at PV onset and fibrotic progression, with splenomegaly and leukocytosis also identified as risk factors. Bone marrow biopsy in PV is therefore crucial for accurate diagnosis, especially in ambiguous cases, and provides prognostic information about progression to post-PV myelofibrosis. The recognition of a specific histological bone marrow pattern in PV led to the “promotion” of bone marrow histology to a major diagnostic criterion in the 2016 WHO revision. Bone marrow biopsy is recommended in practical diagnostic algorithms for PV and secondary polycythemia.
Debates persist regarding the applicability and reproducibility of histological criteria for pre-PMF and ET differentiation in routine practice. Some propose more objective, algorithmic approaches including quantitative morphological feature assessment for clearer ET and pre-PMF separation. However, diagnosing early-stage subtypes involves considering the entire complex bone marrow architecture in MPN, best captured by specific diagnostic patterns rather than single parameters. In PMF, bone marrow fibrosis grading significantly impacts clinical presentation and outcome. A major advancement in the 2016 WHO revision was clarifying the incidence and maximum reticulin fibrosis grade in ET to enhance differentiation from PMF. Reproducibility of WHO-defined morphological features for ET and pre-PMF differentiation has been evaluated in large cohorts, achieving >80% diagnostic consensus with interobserver variability assessment. Studies including reactive lesions, normal bone marrow, and MPN subtypes to mimic “real-world” pathology settings showed good reliability in applying WHO guidelines. The proportion of MPN-unclassifiable (MPN-U) cases can reflect diagnostic accuracy in MPN subtyping. MPN-U incidence varies significantly across studies, ranging up to >20%, but most studies report 10-15% or less, with reduced incidence when applying 2016 WHO criteria. These variations may relate to reviewer experience, early-phase MPN cases, prior cytoreductive treatment effects on morphology, and incomplete clinical/mutation data.
Challenges and Future Directions in Applying WHO Criteria
The 2016 WHO revision aims for immediate routine application, particularly in early PV diagnosis and clearer pre-PMF differentiation from ET and overt PMF, given their prognostic significance. Recognizing early and distinct disease phases through homogeneous clinical, histopathological, and molecular patterns improves management and potentially outcomes. For example, CALR mutations are very unlikely in PV, and MPL mutations virtually negate it. Adopting the 2016 WHO criteria as a “state-of-the-art” approach in clinical practice is crucial for collecting homogeneously defined patient categories to assess clinical course, outcome, and treatment response, and for further molecular and cellular studies to discover diagnostic biomarkers. Gene and non-coding RNA expression profiles, NanoString interrogation of bone marrow tissues, inflammatory cytokine/chemokine levels, and cell membrane antigen combinations are promising research areas. These novel techniques may eventually complement or even replace bone marrow biopsies, which currently remain essential in modern MPN diagnosis.
Hematopathologist Perspective: Challenges and Future Directions
Reproducibility of histological characteristics in the WHO classification remains debated. While overall histological evaluation shows high reproducibility, specific morphological feature identification has more limited reproducibility among hematopathologists, with consensus ranging from 49-100% in some studies. Reasons for this include: (1) failure to reproducibly identify standard bone marrow features, (2) small, non-representative biopsy specimens, (3) disregard of age-related cellularity adjustments, (4) inaccurate fiber grading due to staining artifacts, and (5) inexperienced investigators.
Central pathology review may be beneficial in certain clinical settings. A recent study showed low concordance (55.8%) in bone marrow fibrosis grading between local pathologists and central review, contrasting with excellent agreement rates (>80%) in central pathology reviews, which are considered excellent by concordance strength standards. These issues are addressable. Training sessions can significantly improve the weighting of individual features defining morphological patterns. Educational seminars and workshops for hematopathologists can enhance the integration of histological characteristics into reproducible MPN subtyping, including increased consensus on pre-PMF identification.
Distinguishing MPN subtypes from MDS/MPN overlap syndromes or MDS, especially in clonally undefined (triple-negative) PMF, is clinically crucial due to significantly different outcomes.
Conclusions
The WHO committee has provided a comprehensive and practical outline of diagnostic criteria for ET, PV, and PMF. Bone marrow examination at MPN diagnosis and during follow-up for progressive disease is strongly recommended. Sufficient bone marrow aspiration is essential for mutation screening and cytogenetic analysis. Morphology is critical for differentiating ET from pre-PMF and JAK2-mutated ET from PV. These distinctions are prognostically relevant, with survival being longest in WHO-defined ET and significantly worse in pre-PMF and PV. Bone marrow examination is optimal for obtaining cytogenetic information, which influences survival in PMF and PV. Driver mutation status complements morphologic diagnosis and provides prognostic information. PV is almost always JAK2-mutated, but driver mutations alone do not distinguish MPNs. However, higher JAK2V617F allele burden favors mPV over ET. In prognosis, thrombosis risk in ET is linked to JAK2 mutations, while type 1/CALR mutations in PMF indicate better survival. Future myelofibrosis diagnosis and MPN characterization will increasingly incorporate other mutations to refine morphologic diagnoses and enhance prognostic accuracy.
Conflict of Interest
The authors declare no conflict of interest.