Sickle cell anemia, an inherited blood disorder affecting hemoglobin, poses significant physical and psychological challenges to those living with it. For nurses, a deep understanding of this condition is crucial to provide effective, patient-centered care. Our role as advocates and providers of holistic care is vital in supporting individuals with sickle cell anemia throughout their lives.
This article delves into sickle cell anemia from a nursing perspective, focusing on the critical aspect of nursing diagnoses. We will explore the pathophysiology, clinical manifestations, and evidence-based interventions, with a specific emphasis on formulating accurate and effective nursing diagnoses to optimize patient outcomes. By enhancing our knowledge of this complex disease, we can better deliver compassionate care and improve the quality of life for patients affected by sickle cell anemia.
Understanding Sickle Cell Anemia
Sickle cell anemia is a genetic form of hemolytic anemia, characterized by the production of abnormal hemoglobin.
- Sickle cell anemia is defined as a severe hemolytic anemia resulting from inheriting the sickle hemoglobin gene. This genetic condition leads to the production of sickle hemoglobin (HbS), which distorts red blood cells into a sickle shape.
- The sickle hemoglobin (HbS) gene is most commonly inherited in individuals of African descent, and to a lesser extent, in populations from the Middle East, the Mediterranean region, and indigenous tribes in India.
- It’s important to note that sickle cell anemia represents the most severe form of sickle cell disease, a group of inherited blood disorders.
Pathophysiology of Sickle Cell Anemia
The root of sickle cell anemia lies in a genetic mutation that affects the hemoglobin molecule.
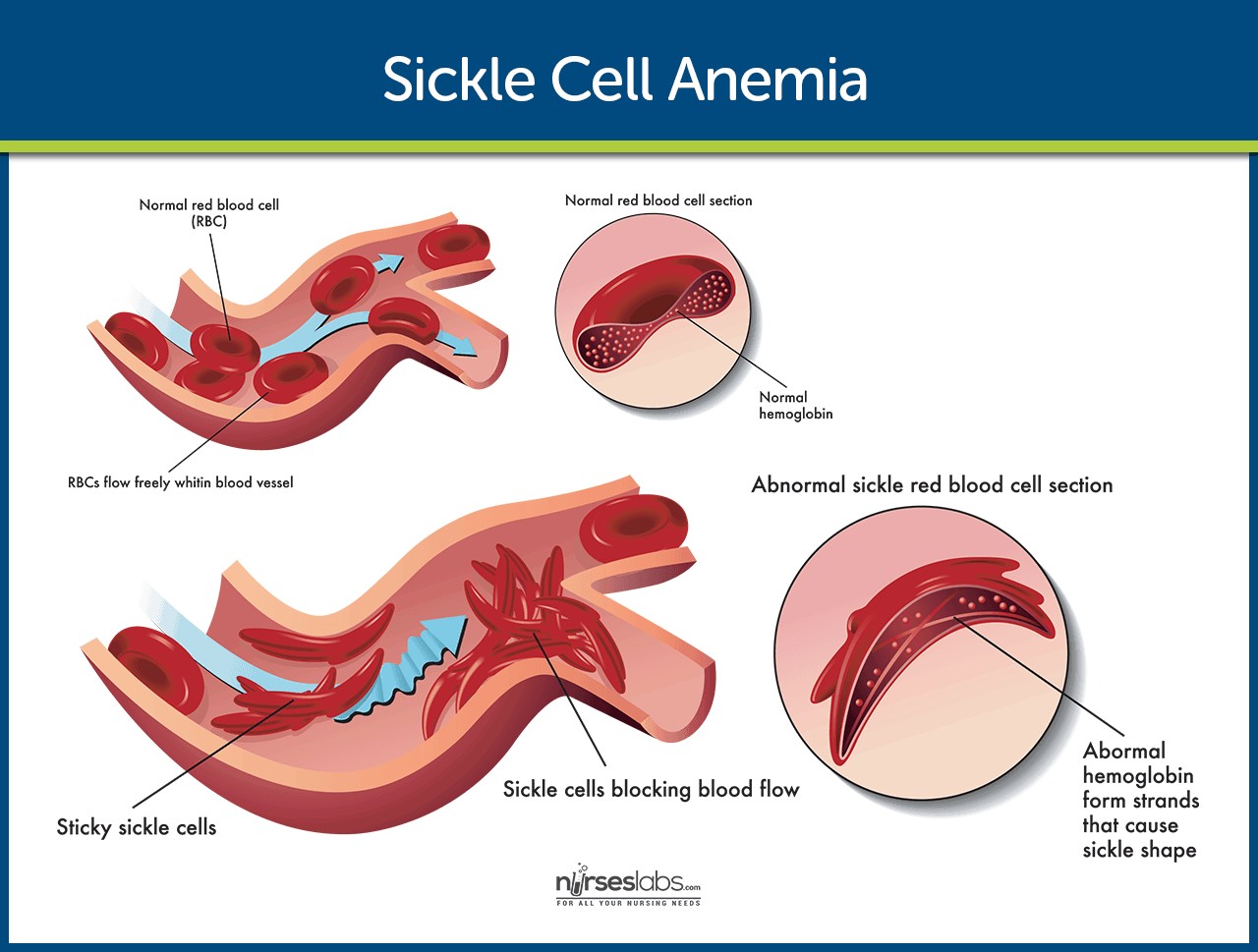
Normal RBC vs Sickled RBC: Illustration comparing the shape of a normal red blood cell to a sickled red blood cell in sickle cell anemia.
The HbS gene causes a critical defect in the hemoglobin molecule. This defect causes red blood cells to become rigid and sickle-shaped, especially when oxygen levels are low. These sickled cells are less flexible than normal red blood cells and can stick to the walls of blood vessels, blocking blood flow. This blockage leads to a variety of complications, including pain crises, organ damage, and anemia. The chronic hemolytic anemia in sickle cell disease arises from the premature destruction of these sickled red blood cells.
Etiology of Sickle Cell Anemia
Sickle cell anemia is primarily caused by genetic inheritance. Specifically, it is caused by inheriting two copies of the sickle cell gene, one from each parent. If an individual inherits only one copy of the gene, they will have sickle cell trait but not sickle cell anemia. Sickle cell trait means they are carriers of the disease and can pass the gene on to their children, but they usually do not experience symptoms of sickle cell anemia themselves.
Clinical Presentation of Sickle Cell Anemia
The symptoms of sickle cell anemia are diverse and vary in severity among individuals. The clinical manifestations are not solely determined by the amount of HbS present but also by other genetic and environmental factors.
- Anemia: Anemia is a constant feature of sickle cell anemia. Patients typically present with hemoglobin levels ranging from 7 to 10 g/dL, reflecting the chronic hemolytic nature of the disease. This reduced oxygen-carrying capacity contributes to fatigue and weakness.
- Jaundice: Jaundice, a yellowing of the skin and sclerae (whites of the eyes), is a hallmark sign. It is caused by the increased breakdown of red blood cells, leading to elevated bilirubin levels.
- Cardiac Dysrhythmias: In adults, the chronic strain on the cardiovascular system can lead to dysrhythmias and even heart failure. The heart works harder to compensate for the reduced oxygen-carrying capacity of the blood.
- Bone Marrow Expansion: In children, the bone marrow attempts to compensate for the chronic anemia by expanding to produce more red blood cells. This can sometimes result in the enlargement of facial and skull bones, which is a characteristic feature in pediatric patients with sickle cell anemia.
Potential Complications of Sickle Cell Anemia
Sickle cell anemia can lead to a wide range of complications due to chronic hemolysis and vaso-occlusion:
- Vaso-occlusive Crisis (Pain Crisis): This is the most common complication, characterized by episodes of severe pain caused by sickled red blood cells blocking blood flow to organs and tissues.
- Acute Chest Syndrome: A life-threatening complication involving lung injury, often mimicking pneumonia.
- Stroke: Sickled cells can block blood vessels in the brain, leading to stroke, especially in children.
- Splenic Sequestration Crisis: The spleen can become enlarged and trap a large volume of red blood cells, leading to a sudden drop in hemoglobin and potentially hypovolemic shock.
- Aplastic Crisis: A temporary cessation of red blood cell production in the bone marrow, often triggered by parvovirus B19 infection.
- Chronic Organ Damage: Long-term vaso-occlusion can damage various organs, including the kidneys, liver, lungs, and heart.
- Pulmonary Hypertension: High blood pressure in the arteries of the lungs, which can lead to heart failure.
- Leg Ulcers: Chronic skin ulcers, particularly on the lower legs, due to poor circulation.
- Priapism: Prolonged and painful penile erection due to vaso-occlusion in the penis.
- Infection: Increased susceptibility to infections due to splenic dysfunction.
Diagnostic Evaluation in Sickle Cell Anemia
Diagnosis of sickle cell anemia relies on a combination of laboratory tests and clinical findings.
- Complete Blood Count (CBC): A CBC reveals several characteristic abnormalities:
- Reticulocytosis: Elevated reticulocyte count (immature red blood cells), ranging from 30% to 50%, indicating the bone marrow’s attempt to compensate for red blood cell destruction.
- Leukocytosis: Increased white blood cell count, especially during vaso-occlusive crises. Counts exceeding 20,000 may suggest infection.
- Decreased Hemoglobin (Hb): Low hemoglobin levels (5–10 g/dL) and reduced total red blood cell count confirm anemia.
- Elevated Platelets: Increased platelet count.
- Mean Corpuscular Volume (MCV): Usually normal to elevated.
- Stained Red Blood Cell Examination: Microscopic examination of a blood smear shows characteristic sickled, crescent-shaped red blood cells. Other abnormalities include anisocytosis (variation in red blood cell size), poikilocytosis (variation in red blood cell shape), polychromasia (immature red blood cells), target cells, Howell-Jolly bodies, basophilic stippling, and occasional nucleated red blood cells (normoblasts).
- Sickle-Turbidity Tube Test (Sickledex): This rapid screening test detects the presence of hemoglobin S (HbS) but does not differentiate between sickle cell anemia and sickle cell trait.
- Hemoglobin Electrophoresis: This definitive test identifies abnormal hemoglobin types and distinguishes between sickle cell trait and sickle cell anemia. Results may be inaccurate if the patient has received a blood transfusion within 3–4 months prior to testing.
- Erythrocyte Sedimentation Rate (ESR): Typically elevated, indicating inflammation.
- Erythrocyte Fragility: Decreased osmotic fragility (resistance to rupture in hypotonic solutions) and reduced red blood cell survival time due to accelerated breakdown.
- Arterial Blood Gases (ABGs): May show decreased partial pressure of oxygen (PaO2) due to impaired gas exchange in the lungs and acidosis (hypoxemia and acidic states during vaso-occlusive crises).
- Serum Bilirubin (Total and Indirect): Elevated due to increased red blood cell hemolysis.
- Acid Phosphatase (ACP): Elevated due to the release of erythrocytic ACP into the serum.
- Alkaline Phosphatase: Elevated during vaso-occlusive crises, indicating bone and liver damage.
- Lactate Dehydrogenase (LDH): Elevated due to red blood cell hemolysis.
- Serum Potassium and Uric Acid: Elevated during vaso-occlusive crises due to red blood cell hemolysis.
- Serum Iron: May be elevated or normal due to increased iron absorption from excessive red blood cell destruction.
- Total Iron-Binding Capacity (TIBC): Normal or decreased.
- Urine/Fecal Urobilinogen: Increased, representing more sensitive indicators of red blood cell destruction than serum levels.
- Intravenous Pyelogram (IVP): May be performed to assess kidney damage.
- Bone Radiographs: Can reveal skeletal changes such as osteoporosis, osteosclerosis, osteomyelitis, or avascular necrosis.
- X-rays: May indicate bone thinning and osteoporosis.
Medical Interventions for Sickle Cell Anemia
Medical management of sickle cell anemia is continually evolving with ongoing research.
- Peripheral Blood Stem Cell Transplant: This is the only potentially curative treatment for sickle cell anemia. However, it is limited to a small subset of patients due to the need for a compatible donor and contraindications such as severe organ damage.
- Transfusion Therapy: Chronic red blood cell transfusions can be effective in preventing or managing complications of sickle cell anemia by reducing the proportion of sickle hemoglobin.
- Supportive Therapy: Supportive care is crucial, especially during painful sickling episodes.
- Hydration: Adequate hydration is paramount to reduce blood viscosity and prevent further sickling.
- Pain Management: Aspirin and NSAIDs are useful for mild to moderate pain. Opioid analgesics may be necessary for severe pain crises.
- Pulmonary Function Monitoring and Management: Regular monitoring of pulmonary function is essential, and pulmonary hypertension should be treated promptly if detected.
- Infection Prevention and Management: Prompt treatment of infections and acute chest syndrome is crucial, as these conditions can trigger sickle cell crises.
- Incentive Spirometry: Recommended to prevent pulmonary complications.
- Bronchoscopy: May be used to identify the source of pulmonary disease.
Pharmacological Agents
- Hydroxyurea: This medication has proven effective in increasing fetal hemoglobin levels, which can reduce the severity of sickle cell anemia symptoms.
- Arginine: Arginine possesses antisickling properties and enhances nitric oxide availability, a potent vasodilator. This can help reduce pulmonary artery pressure and improve blood flow.
Nursing Management: A Patient-Centered Approach
Nursing Management: Illustrative image representing the nursing process, crucial for managing patients with sickle cell anemia.
Nursing management is integral to the comprehensive care of patients with sickle cell anemia. It encompasses a thorough nursing assessment, accurate nursing diagnoses, meticulous care planning, effective interventions, and continuous evaluation of patient outcomes.
Comprehensive Nursing Assessment
A thorough nursing assessment is the foundation of effective care for patients with sickle cell anemia. Key assessment areas include:
- History of Sickle Cell Crises: Elicit information about factors that have triggered previous crises, as well as the patient’s usual strategies for prevention and management.
- Pain Assessment: Regularly assess pain levels using a validated pain intensity scale.
- Pain Characteristics: Determine the quality, frequency, location, and factors that exacerbate or alleviate pain.
- Infection Assessment: Monitor for signs and symptoms of infection, as patients with sickle cell anemia are highly susceptible to infections.
Nursing Diagnoses for Sickle Cell Anemia
Based on the nursing assessment data, several nursing diagnoses may be appropriate for patients with sickle cell anemia. These diagnoses guide the development of individualized care plans. Prioritizing nursing diagnoses is crucial based on the patient’s immediate needs and presenting symptoms.
Common Nursing Diagnoses for Sickle Cell Anemia:
- Acute Pain related to vaso-occlusive crisis as evidenced by patient report of severe pain, guarding behavior, and elevated vital signs.
- Fatigue related to decreased oxygen-carrying capacity secondary to anemia as evidenced by patient report of exhaustion, weakness, and inability to perform usual activities.
- Risk for Infection related to impaired splenic function and chronic illness.
- Ineffective Tissue Perfusion (Peripheral) related to vaso-occlusion and sickled red blood cells as evidenced by delayed capillary refill, pain, and skin color changes.
- Deficient Fluid Volume related to inadequate fluid intake and increased fluid loss during vaso-occlusive crisis.
- Activity Intolerance related to fatigue and pain as evidenced by reports of weakness and dyspnea on exertion.
- Anxiety related to chronic illness, pain crises, and potential complications as evidenced by restlessness, irritability, and expressed concerns about prognosis.
- Ineffective Coping related to chronic illness and recurrent pain crises as evidenced by frustration, anger, and difficulty adhering to treatment regimen.
- Deficient Knowledge related to sickle cell anemia management and prevention of crises as evidenced by questions about self-care and lack of understanding of disease process.
- Risk for Powerlessness related to chronic illness and unpredictable pain crises.
- Disturbed Body Image related to physical manifestations of sickle cell anemia, such as jaundice or skeletal deformities.
Prioritizing Nursing Diagnoses:
In acute care settings, Acute Pain and Ineffective Tissue Perfusion (Peripheral) often take priority during a vaso-occlusive crisis. Risk for Infection is also a high priority due to the increased vulnerability of these patients. In chronic management, Fatigue, Anxiety, Ineffective Coping, and Deficient Knowledge become more central to long-term care planning.
Nursing Care Planning and Patient Goals
Main Article: 6 Sickle Cell Anemia Crisis Nursing Care Plans
The overarching goals of nursing care for patients with sickle cell anemia are:
- Effective pain management and relief.
- Reduction in the frequency and severity of sickle cell crises.
- Enhancement of self-esteem and sense of control.
- Prevention and absence of complications.
Nursing Interventions: Implementing Patient Care
Nursing interventions are tailored to address the identified nursing diagnoses and achieve patient goals.
Managing Pain:
- Administer prescribed analgesics promptly and effectively, utilizing a multimodal approach including opioids, NSAIDs, and non-pharmacological pain relief measures.
- Assess pain regularly and document pain intensity, location, quality, and relieving/aggravating factors.
- Employ non-pharmacological pain management techniques such as heat or cold application, massage, relaxation techniques, and distraction.
- Maintain patient comfort and promote rest.
Preventing and Managing Infection:
- Monitor for signs and symptoms of infection, such as fever, chills, increased white blood cell count, and localized signs of infection.
- Implement meticulous infection control measures, including hand hygiene and aseptic technique for invasive procedures.
- Administer prophylactic antibiotics as prescribed.
- Educate patients and families about infection prevention strategies, such as avoiding crowds during cold and flu season and practicing good hygiene.
Promoting Coping Skills:
- Foster a therapeutic nurse-patient relationship based on trust and empathy.
- Focus on the patient’s strengths and coping mechanisms to enhance self-efficacy.
- Provide opportunities for patient decision-making regarding their daily care to promote a sense of control.
- Offer emotional support and encourage verbalization of feelings and concerns.
- Refer patients to support groups and counseling services as needed.
Increasing Knowledge:
- Educate patients about sickle cell anemia, its pathophysiology, and potential complications.
- Teach patients about factors that can precipitate a sickle cell crisis, such as dehydration, infection, extreme temperatures, and stress.
- Instruct patients on preventative measures, including maintaining adequate hydration, avoiding extreme temperatures, managing stress, and seeking prompt medical attention for infections.
- If hydroxyurea is prescribed for women of childbearing age, provide thorough education about the drug’s teratogenic effects and the importance of pregnancy prevention.
Monitoring and Managing Potential Complications:
- Implement management strategies for potential complications as delineated in previous sections.
- Leg Ulcers: Protect legs from trauma and contamination, utilize aseptic technique for wound care, and consult with a wound-ostomy-continence nurse.
- Priapism: Instruct patients to empty the bladder, exercise, and take a warm bath at the onset of an episode. Advise patients to seek immediate medical attention if priapism persists for more than 3 hours.
- Chronic Pain and Substance Abuse: Emphasize adherence to the prescribed treatment plan, promote trust through effective acute pain management, and advocate for continuity of care with a single provider.
Promoting Home and Community-Based Care:
- Involve patients and families in comprehensive education about the disease, treatment, assessment, and monitoring for complications.
- Teach patients and families about vascular access device management and chelation therapy if applicable.
- Encourage regular communication between healthcare providers, patients, and families.
- Provide clear guidelines on when to seek urgent medical care.
- Ensure follow-up care for patients with vascular access devices.
Evaluation of Patient Outcomes
Expected patient outcomes indicate the effectiveness of nursing care. These outcomes are:
- Pain relief and effective pain management.
- Reduced frequency of sickle cell crises.
- Enhanced self-esteem and sense of control.
- Absence of complications or effective management of unavoidable complications.
Discharge Planning and Home Care Guidelines
Patient and family education are paramount for successful home care management.
- Vascular Access Management: Provide training on vascular access device care for patients requiring ongoing infusions at home.
- Communication: Emphasize the importance of ongoing communication among all healthcare providers, patients, and families to ensure coordinated and comprehensive care.
Documentation in Nursing Care
Accurate and thorough documentation is essential for quality nursing care. Key documentation elements for patients with sickle cell anemia include:
- Patient’s description of pain and response to pain management interventions.
- Acceptable pain level achieved.
- Prior medication use and effectiveness.
- Signs and symptoms of infection and interventions implemented.
- Plan of care and any revisions.
- Patient and family teaching provided and their understanding.
- Patient responses to all nursing interventions and treatments.
- Progress towards desired patient outcomes.
- Long-term care needs and discharge planning.
See Also
Related resources for further reading:
- Anemia
- Risk for Infection
- 6 Sickle Cell Anemia Crisis Nursing Care Plans