Abstract
Human cytomegalovirus (CMV) remains a significant contributor to morbidity and mortality, especially in transplant recipients, and is the primary viral cause of congenital infections globally. Current clinical diagnosis of CMV infection and reactivation relies on real-time quantitative polymerase chain reaction (RT-qPCR), which, while effective, suffers from limitations in sensitivity and standardization across different laboratories. A rapid, point-of-care diagnostic tool for CMV viremia is crucial to address these shortcomings and improve patient management, particularly in vulnerable populations. This study introduces the application of clustered regularly interspaced short palindromic repeats (CRISPR)-Cas12a technology for the development and validation of a novel rapid diagnostic assay for CMV. We evaluated this system using human saliva and urine samples spiked with CMV, achieving a sensitivity of approximately 10 infectious units (IU)/mL and demonstrating robust specificity against other herpesviruses. Our findings highlight the potential of this CRISPR-Cas12a-based assay as a point-of-care screening tool in both transplant and neonatal settings, offering a significant advancement over existing diagnostic methods for congenital CMV infection diagnosis.
Keywords: Cytomegalovirus, CMV, CRISPR-Cas12a, gRNA, Rapid diagnostic, Point of Care, Congenital CMV Infection Diagnosis
1. Introduction
Cytomegalovirus (CMV), a double-stranded DNA betaherpesvirus, exhibits a high seroprevalence, affecting 50–80% of adults in the United States and over 95% in developing nations (Cannon et al., 2010; Bate et al., 2010). While primary CMV infection is typically managed by the immune system in immunocompetent individuals, the virus persists for life, with periodic reactivations indicated by detectable viral DNA or CMV-specific IgM or IgG in blood, especially with increasing age (Parry et al., 2016; van Boven et al., 2017). Despite available antiviral treatments, CMV remains a major concern in transplant medicine and is the leading infectious cause of congenital conditions (Dollard et al., 2007). The standard diagnostic method for CMV detection in bodily fluids is real-time quantitative polymerase chain reaction (RT-qPCR). Although considered the gold standard, RT-qPCR has notable drawbacks, including moderate sensitivity (around 100–400 copies/mL) (altona-diagnostics. RealStar CMV PCR Kit, 2017; testguide.labmed.uw.edu. CMV Quantitative by PCR, 2022) and inconsistent standardization across testing facilities, despite the implementation of a World Health Organization (WHO) International Standard in 2010 (Preiksaitis et al., 2016). This lack of standardization leads to variations in preemptive therapy thresholds among different medical centers (Preiksaitis et al., 2016; Hayden et al., 2017). Furthermore, RT-qPCR requires specialized equipment and trained personnel, and the turnaround time for results is typically 24–48 hours (testguide.labmed.uw.edu. CMV Quantitative by PCR, 2022; Aruplab. Cytomegalovirus by Quantitative PCR, 2022).
In immunosuppressed patients, such as transplant recipients, CMV reactivation can lead to significant viremia (Vallejo et al., 2022; Duan et al., 2022). Post-transplantation CMV reactivation is directly linked to severe end-organ diseases like pneumonia, colitis, and retinitis, and is associated with reduced overall survival and increased non-relapse mortality (Teira et al., 2016). In solid organ transplantation, CMV poses a high risk of complications, particularly in the initial 3–6 months post-transplant due to immunosuppressive treatments (Fishman, 2007). Antiviral therapies are available but are limited by potential toxicities and the development of antiviral resistance (Krosky et al., 1998; Imlay and Kaul, 2021). Therefore, rapid and quantitative monitoring of CMV viremia is essential to minimize these treatment-related issues in transplant settings.
Congenital CMV (cCMV) infection is the primary infectious cause of fetal loss, neurological disorders in infants, and congenital disabilities (Zhang et al., 2022; Li et al., 2021). The global birth prevalence of cCMV is estimated at approximately 0.7% (Kenneson and Cannon, 2007; Fowler and Boppana, 2006). Prenatal CMV antibody screening is available for pregnant women with suspected CMV infection, but its clinical utility remains debated (Leruez-Ville et al., 2020). Suspicion of cCMV may arise during fetal imaging if intracranial calcifications or intrauterine growth restriction are detected (Ito et al., 2013). Suspected cCMV cases are then referred to specialists who typically confirm the diagnosis via PCR detection of CMV in amniotic fluid obtained through amniocentesis (Leruez-Ville et al., 2020; Rawlinson et al., 2017). However, amniocentesis carries risks to the pregnancy. Notably, only about 10% of cCMV infections are symptomatic at birth, leading to neurological sequelae (Boppana and Fowler, 2017). In asymptomatic cases, 10–15% of infants may still develop progressive sensorineural hearing loss and cognitive impairments later in life (Fowler and Boppana, 2018; Foulon et al., 2019). Recent studies emphasize the importance of early antiviral intervention to improve hearing and developmental outcomes in symptomatic cCMV neonates (Kimberlin et al., 2015; Nishida et al., 2016; Yamada et al., 2020). Neonatal cCMV detection is confirmed by RT-qPCR in urine or saliva within the first three weeks of life. However, this testing is often initiated based on clinical suspicion, resulting in up to 90% of cCMV cases being missed at birth (Sorichetti et al., 2016). Universal newborn screening is therefore essential to identify the majority of cCMV infections and facilitate timely intervention for improved outcomes.
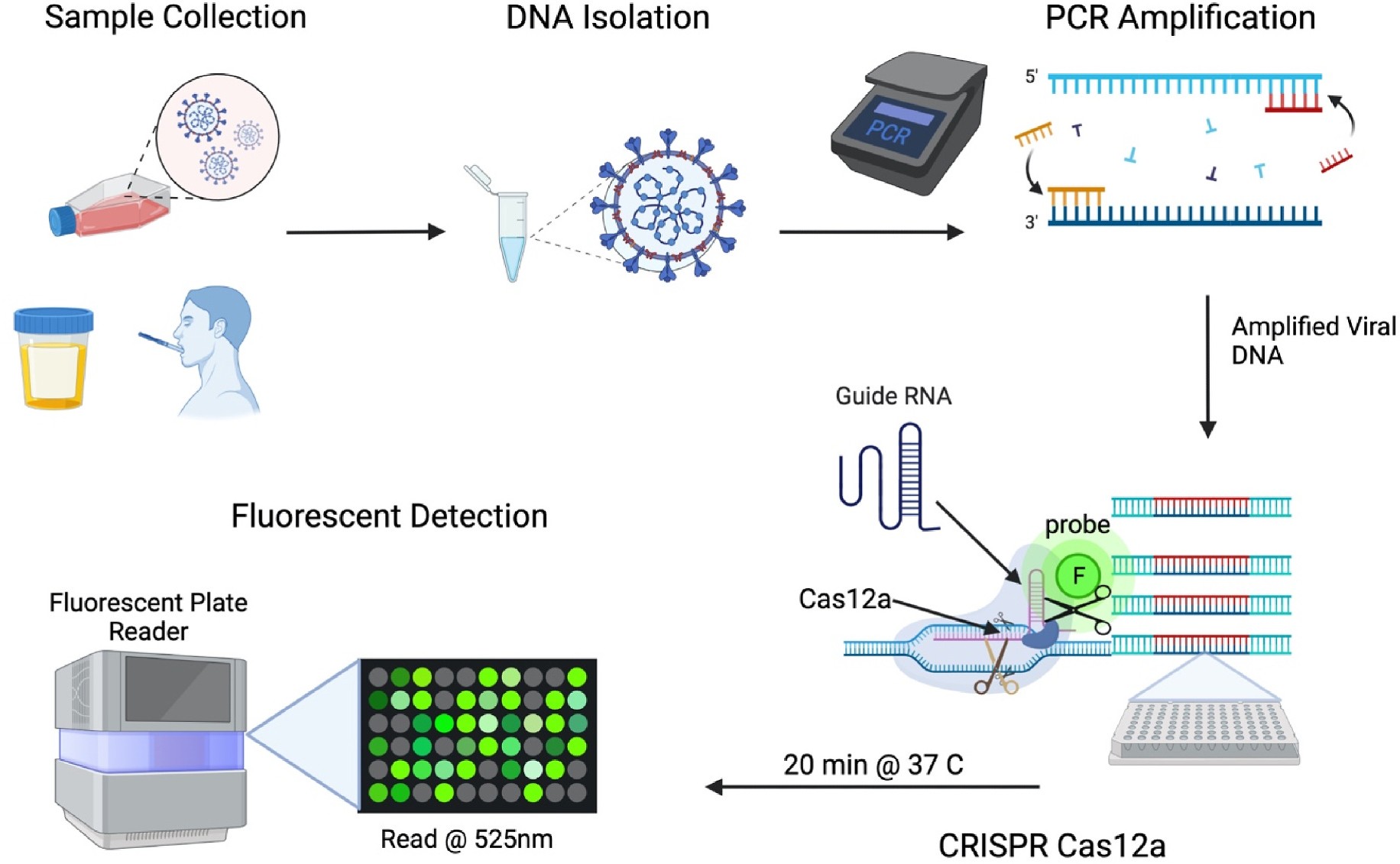
Figure 1: Schematic representation of the CRISPR-Cas12a assay process for rapid CMV detection, highlighting the steps from sample collection to result readout.
The Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) and CRISPR-associated protein (Cas) system is emerging as a promising tool for pathogen detection (Huang et al., 2021; Xu et al., 2020; Kaminski et al., 2020a). CRISPR sequences, consisting of repeat and spacer elements, can be designed to target specific viral DNA sequences. The Cas protein possesses helicase and nuclease activities that cleave DNA near the CRISPR-Cas locus. The CRISPR-Cas system offers high specificity and sensitivity for nucleic acid detection. Specifically, CRISPR-Cas12a recognizes thymine-rich double-stranded DNA protospacer adjacent motifs (PAM). The staggered DNA cleavage by Cas12a provides enhanced target-specific DNA assembly compared to traditional restriction enzyme methods (Kim et al., 2017). Cas12a cleaves DNA 18–24 base pairs downstream of the PAM site, enabling sequence recognition and repeated cleavage events post-repair. This characteristic makes Cas12a suitable for diagnostic applications, where a guide RNA (gRNA) directs Cas12a to the target sequence, and the enzyme’s collateral cleavage activity is used to cleave a fluorescent reporter probe, allowing for quantifiable detection. This principle is central to our assay design for CMV, building upon previous research in the field (Huang et al., 2021; Broughton et al., 2020; Nguyen et al., 2020). An overview of our assay design is illustrated in Figure 1. The urgent need for a low-cost, point-of-care rapid diagnostic for CMV is critical to transform the approach to CMV-related disease management. This study details the design, optimization, and validation of a CRISPR-Cas12a-based rapid diagnostic for CMV, with a focus on its potential for point-of-care congenital CMV infection diagnosis.
2. Methods
2.1. gRNA and Primer Design
To design guide RNAs (gRNAs) for CMV detection, we utilized the CMV Merlin strain sequence (GenBank: AY446894.2). We scanned the CMV genes UL123 and US28 for CRISPR-Cas12a PAM sequences, specifically TTTV (where V represents any base except T). The gRNA sequences were defined as the 20–24 base pairs immediately following the PAM sequence. CMV Merlin-specific primer sets were designed using NCBI Primer-BLAST. Primer properties, including GC content, melting temperature, GC clamp, potential for dimer formation, and hairpin structures, were analyzed using Premier qOligo software biosoft.com. All primers and gRNAs were synthesized by Integrated DNA Technologies (IDT).
2.2. Viral DNA Propagation and Isolation
CMV Towne (ATCC VR-977), Toledo (gifted by Dr. John Sinclair), and TR (gifted by Dr. Andrew Yurochko) strains were propagated in Human Foreskin Fibroblasts (HFF) (ATCC SCRC-1041) using DMEM (Gibco) supplemented with 10% FBS and 1X Glutamax (Gibco). HHV-6A strain HST (obtained from the HHV-6 Foundation) was cultured in HSB-2 cells (ATCC CCL-120.1) in IMDM media (Gibco) with 10% FBS. HHV-6B strain Z29 (from the HHV-6 Foundation) was propagated in Molt-3 cells (ATCC CRL-1552) using RPMI-1640 media (Gibco) with 10% FBS. Human simplex virus 1 (HSV-1) (gifted by Dr. Shitao Li) was propagated in Vero cells (ATCC CCL-81) in DMEM with 10% FBS. All viral strains were purified by centrifugation and titered as previously described (Britt, 2010; Combs et al., 2019). Viral samples (100 μl) were heat-inactivated at 60 °C for 30 minutes and stored at −80 °C. Viral DNA was extracted using the Quick-DNA/RNA Viral Kit (Zymo Research) according to the manufacturer’s instructions and stored in nuclease-free water at −20 °C until use.
2.3. Human Samples
Human saliva and urine samples from three healthy donors were kindly provided by Dr. Elizabeth Norton. These de-identified samples (Tulane IRB#15–727936), collected between 2015 and 2016, were aliquoted and stored at −80 °C. Samples were thawed, and 90 μL aliquots were spiked with 10 μL of titered heat-inactivated CMV Towne, TR, or Toledo strains, resulting in CMV concentrations ranging from 105 to 10-1 viral IU/mL. DNA/RNA Shield (Zymo Research) was added to the samples, and DNA was extracted using the Quick-DNA/RNA Viral Kit (Zymo Research).
2.4. PCR
PCR reactions were performed for each viral target region in a 20 μL volume, containing 5 μL of DNA template and 15 μL of PCR mastermix (0.5 μL forward primer [10 μM], 0.5 μL reverse primer [10 μM] [IDT], 0.2 μL AccuPrime Taq DNA Polymerase, 2 μL AccuPrime PCR Buffer [10x] [ThermoFisher], and 11.8 μL nuclease-free water [Invitrogen]). DNA amplification was carried out using a T100 thermocycler (Bio-Rad) with the following cycling conditions: initial denaturation at 94 °C for 2 minutes, followed by 38 cycles of denaturation at 98 °C for 10 seconds, annealing at 60 °C for 10 seconds, and extension at 72 °C for 15 seconds, with a final elongation step at 72 °C for 5 minutes. Amplified PCR products were used immediately or stored at −20 °C.
2.5. CRISPR-Cas12a Fluorescent Detection Assay
The CRISPR-Cas12a fluorescent detection assay was performed under previously optimized conditions, adjusting the molar ratio of Cas12a/gRNA to fluorescent probe to 1:3, modified from a 1:7.5 ratio used for RNA virus detection (Huang et al., 2020). Each 32 μL reaction contained final concentrations of 2 μL RT-PCR product, 10 μM gRNA, 6.7 μM oligonucleotide fluorescent probe (IDT), 10 μM LbCas12a (New England Biolabs), 3 μL 10x NEBuffer 2.1 (New England Biolabs), and 11.8 μL nuclease-free water. Reactions were performed in triplicate in a 96-well opaque black flat-bottom microplate (Corning) and incubated in the dark at 37 °C for 20 minutes. Fluorescence was measured at 525 nm every 5 minutes using a Synergy H1 Microplate reader (BioTek). A positive cutoff value was determined as the mean fluorescence of the no template control (NTC) plus three times the standard deviation (SD) (3*SD).
2.6. Specificity and Cross-reactivity Testing
In silico specificity testing was conducted using the NCBI BLAST tool to assess the gRNA/primer sets for potential cross-reactivity with organisms other than CMV strains in the non-redundant nucleotide database. In vitro specificity was evaluated by testing HSV-1, HHV-6A (strain HST), and HHV-6B (strain Z29) against each CMV-targeted gRNA/primer set. gRNA/primer sets showing cross-reactivity (defined as a positive value exceeding the NTC mean + 3*SD) were excluded from further analysis.
2.7. Limit of Detection Determination
The limit of detection (LoD) for gRNA/primer sets without cross-reactivity was determined using serial dilutions of isolated viral DNA. Dilutions of purified and spiked viral DNA, ranging from 105 to 10-1 IU/mL for each CMV strain and sample type, were tested using the CRISPR-Cas12a assay. A sample was considered positive if its fluorescence value was greater than the mean of the NTC plus 3*SD. This method was used to determine the lowest detectable viral DNA dilution for each gRNA/primer set and sample type/viral strain. The LoD testing was performed in triplicate using samples from three different biological donors (as described in section 2.3).
2.8. Statistical Analysis
Data are presented as means and standard deviations. One-way ANOVA was used to compare intensity responses for sensitivity and specificity thresholds across different CMV strains. Dunnett’s multiple comparison test was used for post-hoc analysis. Statistical analyses were performed using GraphPad Prism (Version 9, GraphPad Software Inc.), with a type I error threshold of 5%. Figures were created using BioRender.com.
3. Results
3.1. Limit of Detection of CRISPR-Cas12a CMV Rapid Diagnostic
Using the CMV Merlin strain genome as a reference, we identified PAM sequences and designed gRNAs targeting the UL123, UL122, and US28 gene regions (Fig. 2). To assess the detection capability of the CRISPR-Cas12a system, CMV was spiked into nuclease-free water at concentrations from 105 to 10-1 infectious units (IU)/mL. Viral DNA was extracted from these spiked samples, and the limit of detection was evaluated. For CMV TR strain, the assay demonstrated significant detection down to 10 IU/mL (Fig. 3A), while for Toledo and Towne strains, the limit of detection was 100 IU/mL (Fig. 3B and C). Notably, the UL123 target achieved a limit of detection as low as 0.1 IU/mL with CMV TR and Toledo strains. CMV Towne was detectable at 10 IU/mL using the UL123 target. The US28 target showed similar sensitivity, with detection limits of 0.1 IU/mL for TR, Toledo, and Towne strains (Fig. 3D and E), and achieved statistical significance at 100 IU/mL for TR, 10 IU/mL for Toledo, and 1 IU/mL for Towne. In contrast, we were unable to detect UL122 across all CMV strains tested (Supplemental Figs. 1A–C). These findings confirm the successful design and functionality of the CRISPR-Cas12a system for CMV detection under controlled laboratory conditions.
Fig. 2. Primer and guide RNA (gRNA) sequences for CRISPR-Cas12a detection of CMV.
Figure 2: Detailed sequences of forward and reverse primers and guide RNAs (gRNAs) designed for CRISPR-Cas12a detection of CMV UL123, US28, and UL122 gene targets, including the identified PAM sequences and primer design strategy.
Fig. 3. Limit of Detection using a CRISPR-Cas12a CMV assay.
Figure 3: Graphs illustrating the limit of detection for the CRISPR-Cas12a CMV assay across different CMV strains and gene targets. (A-C) Detection limits for UL123 gene using CMV strains TR, Toledo, and Towne, respectively. (D-F) Detection limits for US28 gene using CMV strains TR, Toledo, and Towne, respectively. Data points represent mean fluorescence values, and dotted lines indicate the positive cutoff threshold.
3.2. Specificity Validation of the CRISPR-Cas12a Assay
To evaluate the viral specificity of the CRISPR-Cas12a CMV rapid diagnostic, we tested its ability to differentiate CMV from other closely related human herpesviruses. As shown in Fig. 4A, the CMV UL123 target was effectively detected in CMV strains TR, Toledo, and Towne, but not in the alphaherpesvirus human simplex virus 1 (HSV-1) or the betaherpesviruses human herpesvirus 6A (HHV-6A) and HHV-6B. Similar specificity was observed using US28 targets (Fig. 4B). Positive detection was defined as fluorescence values exceeding the NTC mean + 3*SD. These results demonstrate that the CRISPR-Cas12a CMV rapid diagnostic exhibits high specificity and does not produce false positives with other common herpesviruses.
Fig. 4. Viral specificity of a CRISPR-Cas12a assay.
Figure 4: Specificity analysis of the CRISPR-Cas12a assay for CMV. (A) Specificity testing using UL123 target against CMV strains (Towne, TR, Toledo) and other herpesviruses (HSV-1, HHV-6A, HHV-6B). (B) Specificity testing using US28 target against the same panel of viruses. Graphs show fluorescence signal intensities, with dotted lines indicating the positive cutoff threshold.
3.3. Performance of CRISPR-Cas12a Assay in CMV-Spiked Human Saliva
Given that CMV is abundantly secreted in saliva, we assessed the sensitivity of the CRISPR-Cas12a diagnostic in this clinically relevant fluid. Saliva samples spiked with CMV at concentrations ranging from 105 to 10-1 IU/mL were tested using UL123 and US28 targets. UL123 detection was significant at 0.1 IU/mL for TR and Toledo strains (Fig. 5A and B). For Towne UL123, detection was observed at 0.1 IU/mL, but significance was reached at 100 IU/mL (Fig. 5C). The limit of detection for US28 in saliva was slightly less robust. TR and Toledo strains were detected at 0.1 IU/mL, with significance at 1 IU/mL for Toledo and 100 IU/mL for TR (Fig. 5D and E). Towne strain was detected at 1 IU/mL, but did not reach statistical significance (Fig. 5F). These results indicate that the CRISPR-Cas12a CMV rapid diagnostic can effectively detect CMV in saliva samples without requiring extensive sample preparation, making it a promising tool for non-invasive point-of-care congenital CMV infection diagnosis.
Fig. 5. The limit of detection in saliva using a CRISPR-Cas12a CMV assay.
Figure 5: Limit of detection of the CRISPR-Cas12a CMV assay in spiked saliva samples. (A-C) Detection limits for UL123 target in saliva spiked with CMV strains TR, Toledo, and Towne, respectively. (D-F) Detection limits for US28 target in saliva spiked with CMV strains TR, Toledo, and Towne, respectively. Graphs display fluorescence intensities and positive cutoff thresholds.
3.4. Performance of CRISPR-Cas12a Assay in Spiked Human Urine Samples
CMV detection in urine is clinically important, particularly for diagnosing congenital CMV infection in neonates. Therefore, we evaluated the assay’s sensitivity using CMV-spiked urine samples, targeting UL123 and US28 genes. The UL123 target achieved a limit of detection of 0.1 IU/mL in urine for all CMV strains, with significance levels at 0.1 IU/mL for TR, 100 IU/mL for Toledo, and 1 IU/mL for Towne (Fig. 6A–C). US28 showed similar performance, with a detection limit of 0.1 IU/mL for TR and Towne strains (Fig. 6D and F), and 10 IU/mL for Toledo (Fig. 6E). Significance for US28 in urine was observed at 100 IU/mL for TR, 10 IU/mL for Toledo, and 100,000 IU/mL for Towne. These results confirm the assay’s potential for sensitive CMV detection in urine, a key sample type for point-of-care congenital CMV infection diagnosis.
Fig. 6. Limit of detection in urine.
Figure 6: Evaluation of the CRISPR-Cas12a CMV assay’s limit of detection in spiked urine samples. (A-C) Detection limits for UL123 target in urine spiked with CMV strains TR, Toledo, and Towne. (D-F) Detection limits for US28 target in urine spiked with CMV strains TR, Toledo, and Towne. Graphs show fluorescence intensity and positive cutoff values.
3.5. Rapid Detection Time of CRISPR-Cas12a Assay
Finally, we assessed the speed of CMV detection using the CRISPR-Cas12a assay. Using CMV concentrations from 105 to 10-1 IU/mL, we monitored fluorescence intensity over time. Consistent with the limit of detection findings (Fig. 3), CMV detection at 101 IU/mL was consistently achieved within 15 minutes of adding the amplified sample (Fig. 7A–L) using both UL123 and US28 targets. The total assay time, including DNA extraction (5 min), PCR amplification (30 min), CRISPR-Cas12a detection (15 min), and sample preparation (30 min), is approximately 90 minutes. While strain-specific and target-specific variations were observed, these data support the potential for rapid CMV analysis using CRISPR-Cas12a detection following target amplification. This rapid detection capability further strengthens the assay’s suitability for point-of-care congenital CMV infection diagnosis and other urgent diagnostic needs.
Fig. 7. Rapid detection of CMV products using CRISPR-Cas12a assay.
Figure 7: Real-time fluorescence monitoring of CMV detection using the CRISPR-Cas12a assay. (A-C) Fluorescence intensity progression over 20 minutes for UL123 target with CMV strains TR, Toledo, and Towne. (G-I) Fluorescence intensity progression for US28 target with CMV strains TR, Toledo, and Towne. (D-F) and (J-L) show fluorescence intensity at the 15-minute time point for UL123 and US28 targets, respectively, highlighting the rapid detection capability of the assay.
4. Discussion
The implementation of a robust, quantitative, and cost-effective point-of-care diagnostic for CMV holds significant potential to reduce CMV-associated morbidity and mortality. Rapid diagnostic results and universal screening can prevent or mitigate CMV-related pathologies, thereby improving patient outcomes and alleviating the substantial economic burden associated with CMV diseases, especially in the context of congenital CMV infection diagnosis. In this study, we have successfully designed, optimized, and validated a CRISPR-Cas12a CMV rapid diagnostic assay. The assay was rigorously tested using human saliva and urine, both relevant clinical specimens for CMV detection, particularly in neonatal and transplant settings. Our collective findings pave the way for the development of a rapid diagnostic tool for CMV that can significantly reduce the severity of congenital CMV infections and improve point-of-care congenital CMV infection diagnosis. However, a direct comparative evaluation between this assay and the current gold standard RT-qPCR, using clinical samples from neonates and other patient populations, is necessary before definitive conclusions can be drawn for clinical application.
We validated our rapid diagnostic test using biologically relevant samples. Urine has been established as a reliable indicator of congenital CMV infection. Studies have shown that infant urine samples exhibit significantly higher viral loads compared to umbilical cord blood or cerebrospinal fluid (CSF) (Halwachs-Baumann et al., 2002), suggesting that urine is highly representative of early CMV infection. Furthermore, challenges in isolating CMV from plasma have been previously noted (Kaminski et al., 2020b), making urine a more accessible and reliable sample type. Urine collection is non-invasive, simple, and does not require specialized medical training, making it ideal for universal CMV screening in healthcare facilities or even for at-home monitoring. Despite its advantages, urine presents unique analytical challenges, such as PCR inhibition due to high urea concentrations (>50 mM) (Khan et al., 1991). To address this, we tested our assay in urine samples to assess its performance in the presence of potential PCR inhibitors and to evaluate its limit of detection in this complex matrix.
Parallel testing with saliva samples yielded results comparable to previously reported CMV detection sensitivities (Yamamoto et al., 2006; Ross et al., 2014). Prior studies using RT-PCR for congenital CMV infection diagnosis in saliva have reported a limit of detection around 200 copies/mL or did not provide quantitative data (Ross et al., 2014), (eurofins-viracor. Cytomegalovirus, 2022). It is important to note that some studies, such as Ross et al., omitted a viral DNA extraction step (Ross et al., 2014), which might explain their lower sensitivity compared to our findings. Our approach, incorporating a DNA extraction step, provides valuable insights for optimizing the CRISPR-Cas12a assay and progressing towards a true point-of-care diagnostic system. Saliva, with its simple and less invasive collection method compared to blood and urine, is the preferred sample type for point-of-care congenital CMV infection diagnosis applications.
This study has two primary limitations. First, our assay was not directly tested on fresh clinical samples. Clinical validation using a prospective cohort is essential to confirm these findings in real-world diagnostic settings. Second, we did not validate the CRISPR-Cas12a assay with whole blood samples in this study. While saliva and urine are highly relevant for congenital CMV infection diagnosis and neonatal screening, extending the application of this assay to other clinical contexts, such as monitoring CMV reactivation in transplant recipients, would require experimental validation with blood samples. Clinical samples may exhibit lower sensitivity due to the presence of PCR-inhibitory substances that can interfere with DNA amplification (Sidstedt et al., 2018). In future clinical validation studies, we plan to use the WHO International Standard for CMV to standardize our results using international units and to directly compare the performance of our CRISPR-Cas12a assay to current standard-of-care RT-qPCR assays. In the absence of this standardization in the current study, we have reported our results in infectious units/mL, which more accurately reflects the nature of the samples used. However, it is important to acknowledge that determining infectious units/mL relies on viral titering, which can introduce variability and potentially affect the reported limit of detection. Furthermore, similar to PCR-based assays, our CRISPR-Cas12a assay does not differentiate between infectious virus and viral DNA. This distinction is critical when interpreting results from patients on antiviral therapies like letermovir, who may have detectable DNAemia without active infectious virus replication. Understanding these assay limitations across a spectrum of clinical samples will guide future improvements and refinement of specificity and limit of detection. We are currently undertaking validation of the CRISPR-Cas12a system using transplant patient samples, directly comparing our rapid diagnostic results to those obtained with standard-of-care RT-qPCR. A key area for further development is eliminating the in vitro nucleic acid amplification step, which is currently part of our protocol. While our assay significantly reduces the detection time, the inclusion of PCR amplification means it is not yet a true amplification-free point-of-care diagnostic. We are actively exploring methods to remove this amplification step from the protocol to realize the full potential of this diagnostic as a rapid, point-of-care solution for congenital CMV infection diagnosis.
In transplant settings, the current standard of care for CMV management involves blood draws and RT-qPCR analysis to monitor CMV viremia. Antiviral treatment adjustments are made based on these results, which typically take 24-48 hours to return. A point-of-care assay that provides immediate results could enhance antiviral therapy efficacy, shorten treatment durations, and mitigate concerns related to antiviral toxicity and resistance. Current commercial CMV diagnostic kits report in vitro limits of detection around 0.238 International Units/μL (approximately 400 copies/mL) (altona-diagnostics. RealStar CMV PCR Kit, 2017), with many available tests exhibiting limits of detection of 100 copies/mL or higher (Aruplab. Cytomegalovirus by Quantitative PCR, 2022). Some assays claim to detect as low as 10 copies/mL, but often at the cost of reduced specificity (around 94%) (Waggoner et al., 2012; fda.gov. Abbott RealTime CMV, 2017). A PCR-based assay targeting the CMV UL123 region reported a limit of detection of 300 International Units/mL (534 copies/mL) (testguide.labmed.uw.edu. CMV Quantitative by PCR, 2022). Clinical validation of our CRISPR-Cas12a assay using blood samples could potentially enable its integration with existing diagnostic workflows, offering improved specificity and limit of detection compared to current methods.
The primary objective of this study was to provide a proof-of-concept for utilizing CRISPR-Cas12a technology as a rapid diagnostic for CMV, particularly in the context of point-of-care congenital CMV infection diagnosis. We observed some unexpected results during our investigations. Despite using a consistent design approach for CRISPR-Cas12a targets, we found considerable variability in assay performance across different targets. The reason for the complete lack of signal with the UL122 target remains unclear, and we are still investigating the factors contributing to the sensitivity differences between UL123 and US28 targets. Ongoing research is focused on elucidating the biochemical factors that influence CRISPR-Cas12a assay design and performance. One advantage of the CRISPR-Cas12a system is its capacity to incorporate multiple targets into a single assay, which could enhance robustness and sensitivity. We are currently exploring additional CMV targets for inclusion in future assay iterations. Alternatively, tailored CMV diagnostic panels could be developed to address specific clinical needs, such as point-of-care congenital CMV infection diagnosis versus transplant monitoring. Our results demonstrate high specificity for CMV, high sensitivity with accurate detection below 100 IU/mL, and rapid detection of CRISPR-Cas12a-tagged targets from pre-amplified products within 20 minutes. These findings, obtained using saliva and urine samples, underscore the promising future of CRISPR-Cas12a technology for point-of-care CMV diagnostics and specifically for improving point-of-care congenital CMV infection diagnosis. Our near-term goals include further optimization and validation of the CRISPR-Cas12a CMV rapid diagnostic, followed by clinical trials to assess its performance in real-world settings.
In summary, this study highlights the significant potential of a CRISPR-Cas12a-based rapid diagnostic for CMV detection in saliva and urine, with particular relevance to point-of-care congenital CMV infection diagnosis. This assay could be adapted for use with other bodily fluids, including blood and ocular fluid, to detect CMV infection across different clinical scenarios. The low cost, ease of use, and quantitative nature of this assay make it suitable for repeated testing during CMV-related complications. We are actively pursuing further validation of this assay in diverse human clinical samples, including neonatal specimens, to demonstrate its feasibility and affordability as a universal neonatal CMV screening tool for improved point-of-care congenital CMV infection diagnosis. Developing an at-home version of this diagnostic is a compelling direction for future research, potentially reducing the need for frequent clinic visits and further enhancing accessibility to rapid CMV testing and point-of-care congenital CMV infection diagnosis.
Supplementary Material
supplemental figure 1
NIHMS1901484-supplement-supplemental_figure_1.pdf (67.1KB, pdf)
Acknowledgements
We gratefully acknowledge the generous support of Dr. Elizabeth Norton for providing urine samples for this study. This work was supported in part by NIH grants 1R21AI169582–01A1 (B.N.), U54GM104940, P20GM103629 (K.J.Z), 1R01HD107790 (K.J.Z), HHV-6 Foundation Pilot Grant (K.J.Z), U54CA260581 (K.J.Z & J.G.S).
Footnotes
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.antiviral.2023.105624.
Data availability
Data will be made available on request.
References
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
supplemental figure 1
NIHMS1901484-supplement-supplemental_figure_1.pdf (67.1KB, pdf)
Data Availability Statement
Data will be made available on request.
References