Thrombotic microangiopathy (TMA) is a serious medical condition characterized by thrombocytopenia, microangiopathic hemolytic anemia (MAHA), and end-organ damage. While seemingly distant from automotive repair, understanding TMA, particularly its diagnosis (“Tma Diagnosis”), can be valuable for professionals in fields requiring a broad knowledge base and problem-solving skills. This review provides an in-depth look at TMA diagnosis, drawing from current medical understanding to create a comprehensive guide.
1. Understanding Thrombotic Microangiopathy (TMA)
Thrombotic microangiopathy (TMA) is not a single disease but rather a pathological process where small blood vessels become blocked by platelet-rich clots. This blockage leads to a reduction in platelets (thrombocytopenia) and the destruction of red blood cells as they squeeze through narrowed vessels (MAHA). This process can affect various organs, leading to significant damage, especially in the kidneys. Prompt and accurate “tma diagnosis” is crucial due to the potentially high morbidity and mortality associated with TMAs. Recent advances in TMA research have led to targeted treatments that have significantly improved patient outcomes, highlighting the importance of staying informed about diagnostic and therapeutic developments in related medical fields.
The clinical presentation of TMA can be diverse, making “tma diagnosis” challenging. Laboratory tests are essential for confirming the diagnosis and identifying the specific type of TMA. The common hallmarks of all TMAs include thrombocytopenia, MAHA, and evidence of organ damage. Thrombocytopenia occurs because platelets are consumed in forming clots. MAHA is indicated by fragmented red blood cells (schistocytes) visible in a peripheral blood smear and elevated lactate dehydrogenase (LDH) levels due to tissue damage and cell breakdown. Haptoglobin, a protein that binds free hemoglobin, is typically low in TMA due to hemolysis. Importantly, the Coombs test, which detects antibodies on red blood cells, is usually negative in TMA, except in pneumococcal hemolytic uremic syndrome. Coagulation tests (PT and aPTT) are typically normal in TMA, which helps to differentiate it from disseminated intravascular coagulation (DIC), where these tests are abnormal.
FIGURE 1.
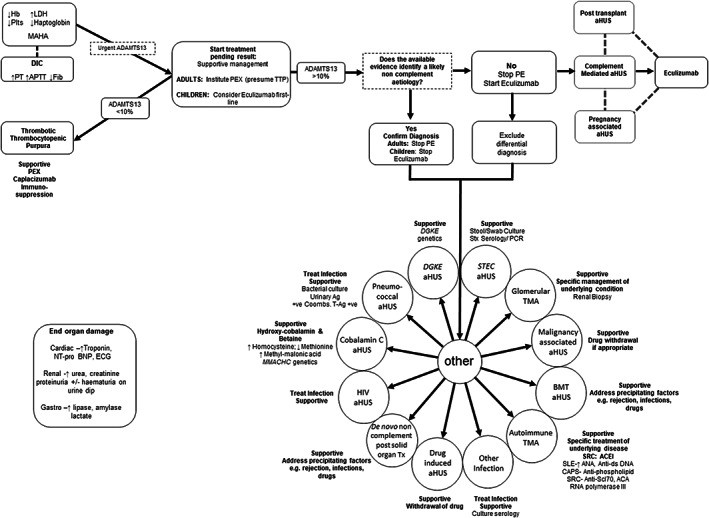 Diagnostic and treatment algorithm for thrombotic microangiopathy (TMA) guiding clinicians from initial presentation with MAHA and thrombocytopenia to identifying the underlying cause and appropriate therapy, emphasizing the critical early consideration of TTP and aHUS.
Diagnostic and treatment algorithm for thrombotic microangiopathy (TMA) guiding clinicians from initial presentation with MAHA and thrombocytopenia to identifying the underlying cause and appropriate therapy, emphasizing the critical early consideration of TTP and aHUS.
Renal involvement is a frequent complication in most TMAs due to the kidney’s dense network of small blood vessels, making it particularly susceptible to damage. While kidney problems are common, TMA can affect other organs as well, and these extra-renal manifestations can vary depending on the specific type of TMA, complicating the “tma diagnosis” process.
Given the potential severity of TMAs, rapid and accurate “tma diagnosis” followed by appropriate treatment is critical for patient survival and long-term well-being. This article will delve into the clinical presentation, diagnostic approaches, and treatment strategies for different types of TMAs, with a strong focus on the diagnostic pathways involved in each.
2. Thrombotic Thrombocytopenic Purpura (TTP): Diagnostic Approach
Thrombotic thrombocytopenic purpura (TTP) is a rare but life-threatening systemic form of TMA. A key factor in “tma diagnosis” for TTP is recognizing its cause: a severe deficiency in the ADAMTS13 enzyme. ADAMTS13 is responsible for cleaving von Willebrand factor (VWF), a protein involved in blood clotting. When ADAMTS13 is deficient, abnormally large VWF multimers accumulate in the blood vessels. These large VWF molecules promote excessive platelet adhesion and aggregation, especially in areas of high blood flow, leading to the formation of clots in small vessels throughout the body.
TTP is uncommon, with an estimated annual incidence of six cases per million in the United Kingdom. TTP can be acquired (aTTP) or congenital (cTTP). Acquired TTP, the more common form in adults, results from autoantibodies that inhibit ADAMTS13 activity. Congenital TTP, more frequent in children and accounting for about 5% of adult cases, is caused by inherited mutations in the ADAMTS13 gene. While ADAMTS13 deficiency is essential for TTP, it is often not the sole trigger. Secondary factors such as pregnancy and infections can initiate the disease process in individuals with ADAMTS13 deficiency, making “tma diagnosis” require consideration of these triggers.
2.1. Clinical Presentation and Laboratory Findings in TTP Diagnosis
Neurological symptoms are prominent in TTP, affecting “tma diagnosis”. Patients may present with headaches, confusion, stroke-like symptoms, or seizures. Other organs frequently involved include the heart and mesentery, leading to cardiac and mesenteric ischemia. Interestingly, kidney involvement, a hallmark of many other TMAs, is less pronounced in TTP. Hematological tests reveal the typical TMA features: MAHA and severe thrombocytopenia, often with platelet counts below 20 × 10^9/L. Cardiac and neurological symptoms are indicators of severe disease and are important for risk stratification in “tma diagnosis”.
TTP is a medical emergency requiring immediate “tma diagnosis” and treatment. Without treatment, mortality can reach as high as 90%. A critical diagnostic test for TTP is the ADAMTS13 activity level. An ADAMTS13 activity level of <10% is highly suggestive of TTP. While specialized labs are needed for definitive ADAMTS13 testing, rapid commercial kits are being developed for faster “tma diagnosis”, although they are not yet widely available. The PLASMIC score is a clinical tool that helps clinicians assess the probability of severe ADAMTS13 deficiency while awaiting test results. Due to the high mortality of TTP, treatment should be initiated based on clinical suspicion, even before ADAMTS13 activity levels are confirmed, emphasizing the urgency of presumptive “tma diagnosis”.
Detecting ADAMTS13 antibodies is also crucial for “tma diagnosis” of acquired TTP. Both ADAMTS13 autoantibody assays and inhibitor assays can be used. Autoantibody assays are preferred as they can detect both inhibitory and non-inhibitory antibodies. Non-inhibitory antibodies, while not directly blocking enzyme activity, can accelerate the clearance of ADAMTS13, contributing to the deficiency. False-negative antibody tests can occur if antibody levels are low or if antibodies are bound in immune complexes, which should be considered in “tma diagnosis” interpretation. ADAMTS13 antigen levels can vary at presentation, but low levels have been associated with poorer outcomes. Cardiac enzyme testing, specifically troponin levels, is important even in the absence of chest pain due to the high incidence of cardiac ischemia in TTP, further aiding in comprehensive “tma diagnosis”.
2.2. aTTP Treatment and Monitoring Post-Diagnosis
2.2.1. Acute Management Following aTTP Diagnosis
Plasma exchange (PE) is the primary treatment for acute acquired TTP (aTTP) following “tma diagnosis”. PE involves removing the patient’s plasma, which contains autoantibodies against ADAMTS13, and replacing it with fresh frozen plasma or cryosupernatant (Octaplas), which provides functional ADAMTS13. PE is continued daily until clinical remission is achieved, defined by a platelet count of ≥150 × 10^9/L and normalized LDH levels, and sustained for at least two days post “tma diagnosis” and initiation of treatment. Many centers use a PE tapering strategy after remission to prevent exacerbations, although the evidence for this is limited, and some studies suggest it might increase complications.
Caplacizumab, an anti-VWF nanobody, is a newer therapy that significantly improves outcomes in aTTP post “tma diagnosis”. Caplacizumab reduces the interaction between large VWF multimers and platelets, thus preventing clot formation. Clinical trials like HERCULES and TITAN, and real-world data, have shown that adding caplacizumab to PE shortens the time to platelet normalization, reduces the duration of PE, and decreases the risk of TTP recurrence post “tma diagnosis”. Clinical exacerbation (recurrence within 30 days of stopping treatment) and relapse (recurrence after 30 days) are important considerations in managing TTP. Caplacizumab is typically started before or with PE and continued daily until 30 days after PE is completed. While effective, caplacizumab is expensive, and research is ongoing to personalize treatment duration based on ADAMTS13 activity monitoring to optimize cost-effectiveness without compromising outcomes post “tma diagnosis”. Studies suggest that monitoring ADAMTS13 activity can guide individualizing caplacizumab treatment duration, potentially shortening it for many patients while ensuring adequate treatment for those needing longer courses. Readily available, rapid ADAMTS13 activity assays would greatly enhance both initial “tma diagnosis” and personalized treatment management. In certain acute aTTP cases, caplacizumab alone without PE has been suggested, but PE plus caplacizumab remains the standard of care following “tma diagnosis”.
Immunosuppression is another key component of acute aTTP treatment after “tma diagnosis”. Standard immunosuppression includes daily prednisolone or pulse methylprednisolone. Rituximab, a monoclonal antibody targeting B cells, is used as a first-line adjunct therapy, particularly in refractory TTP or to reduce relapse risk after remission post “tma diagnosis”.
Refractory TTP, defined as no response after five PE sessions or relapse during standard treatment, requires further management. Integrated analyses of the TITAN and HERCULES trials indicate that caplacizumab reduces the risk of refractory TTP post “tma diagnosis”. In refractory cases, it’s crucial to reassess the “tma diagnosis” and look for contributing factors like infections. Other treatments for refractory TTP include vincristine, bortezomib, azathioprine, and splenectomy.
Patients with aTTP are at increased risk of venous thromboembolism (VTE) and arterial thrombosis. Once the platelet count rises above 50 × 10^9/L, prophylactic low molecular weight heparin and compression stockings are recommended for VTE prevention, unless contraindicated. Antiplatelet therapy is generally not recommended in acute aTTP due to bleeding risks, especially with caplacizumab. Folate supplementation is advised during active hemolysis post “tma diagnosis”.
2.2.2. Follow-up and Long-Term Management After aTTP Diagnosis
Long-term follow-up post “tma diagnosis” is essential for aTTP patients. Regular monitoring of ADAMTS13 activity and intermittent rituximab treatments may be needed to minimize relapse risk.
Open ADAMTS13 is a novel biomarker that can help confirm aTTP, especially when standard autoantibody tests are falsely negative due to antibody clearance. Open ADAMTS13 assays detect conformational changes in ADAMTS13 induced by autoantibody binding, potentially allowing earlier detection of subclinical TTP and prompt treatment initiation, improving long-term management post “tma diagnosis”.
The Oklahoma TTP-HUS Registry has provided valuable insights into long-term outcomes of TTP and HUS patients. Studies from this registry have shown higher rates of cognitive decline and depression in TTP survivors, as well as increased all-cause mortality compared to the general population, highlighting the need for comprehensive long-term care and monitoring post “tma diagnosis”.
2.3. cTTP Treatment Post-Diagnosis
In contrast to aTTP, congenital TTP (cTTP) management post “tma diagnosis” focuses on replacing the deficient ADAMTS13 enzyme rather than immunosuppression. Regular fresh frozen plasma infusions are the mainstay of treatment, providing exogenous ADAMTS13. Recombinant ADAMTS13 (Bax 930) is under clinical investigation as a potential alternative to plasma infusions (NCT03393975), which may simplify long-term management of cTTP post “tma diagnosis”.
3. Complement-Mediated Atypical Hemolytic Uremic Syndrome (aHUS): Diagnostic Pathway
Hemolytic uremic syndrome (HUS) is defined by the triad of thrombocytopenia, MAHA, and acute kidney injury (AKI). Atypical HUS (aHUS) initially broadly encompassed all HUS cases not caused by Shiga toxin-producing bacteria. However, with better understanding of aHUS causes and treatments, the term complement-mediated aHUS is now preferred to specifically describe aHUS cases resulting from dysregulation of the complement system. Accurate “tma diagnosis” must differentiate complement-mediated aHUS from other TMAs.
FIGURE 2.
Pathogenesis of complement-mediated atypical hemolytic uremic syndrome (aHUS). Demonstrates the role of uncontrolled complement activation in aHUS, leading to endothelial damage and TMA, crucial for understanding the therapeutic targets and diagnostic markers.
3.1. Incidence and Pathogenesis of Complement-Mediated aHUS in TMA Diagnosis
Complement-mediated aHUS has an estimated incidence of 0.42 cases per million per year in the United Kingdom. It results from acquired or inherited defects in the alternative pathway (AP) of complement, leading to excessive complement activation and formation of platelet-rich fibrin thrombi, primarily in the renal microvasculature. While kidney involvement is dominant, extra-renal manifestations, particularly neurological, occur in 10%–20% of cases, complicating “tma diagnosis” and requiring a broad differential.
3.2. Complement System Overview for TMA Diagnosis
The complement system is a crucial part of the immune system, comprising over 30 proteins that work to eliminate pathogens and clear immune complexes and damaged cells. It involves three activation pathways: alternative (AP), classical (CP), and lectin (LP), all converging on a common terminal pathway. Understanding the complement system is essential for “tma diagnosis” of aHUS.
The AP is a continuously active amplification loop, boosting immune responses initiated by other pathways. This loop is tightly regulated to prevent excessive activation. Complement C3 undergoes spontaneous hydrolysis to C3a and C3b, with C3b attaching to cell surfaces. On activating surfaces (e.g., bacteria), factor B (FB) binds C3b and is cleaved by factor D to form C3bBb, the AP C3 convertase. This convertase amplifies the cascade by cleaving more C3. With further C3b binding, it becomes a C5 convertase, cleaving C5 into C5a and C5b, initiating the formation of the membrane attack complex (MAC, C5b-9), which lyses cells. C3a and C5a are anaphylatoxins, promoting inflammation. The CP is activated by immune complexes, while the LP is triggered by microbial carbohydrates. Both CP and LP form a different C3 convertase (C4b2a).
3.3. Complement Regulation and Diagnostic Implications in TMA
Factor H (FH) is the main fluid-phase regulator of the AP, competing with FB for C3b, accelerating C3 convertase breakdown, and acting as a cofactor for Factor I (FI). Membrane cofactor protein (CD46) on cell surfaces also acts as an FI cofactor, inactivating C3b and C4b. FI is the central regulatory enzyme, inactivating C3b and C4b with cofactors like FH and CD46. Defects in these regulators are key to complement-mediated aHUS and are targets for “tma diagnosis”.
3.4. Clinical Features and Laboratory Tests for Complement-Mediated aHUS Diagnosis
In complement-mediated aHUS, AKI is typically more prominent than in TTP, aiding in differential “tma diagnosis”. There isn’t a rapid screening test specifically for complement-mediated aHUS. Initial “tma diagnosis” often relies on excluding other TMAs and then confirming complement dysregulation through genetic and autoimmune testing. Routine complement component level tests include C3, C4, FB, FBb, CH50, AH50, FH, FI, C5a, and sC5b9. In aHUS, C3, FB, CH50, and AH50 may be low, while C5a, C5b9, and Bb may be elevated. However, complement profiles alone are not definitive for “tma diagnosis” of complement-mediated aHUS.
Genetic screening for CFH, CFI, C3, CFB, CD46, and CFHR1 is essential to confirm “tma diagnosis” of complement-mediated aHUS. Mutation screening is complex, as many disease-associated mutations are rare and often missense mutations of unknown significance. Serum FI, FH, and cell surface CD46 levels can help assess the pathogenicity of rare genetic variants. In aHUS, >70% of CFI and CD46 variants lead to low protein levels, while CFH variants often result in normal levels of non-functional protein. The CFH gene region is also prone to genomic instability and hybrid gene formation due to low-copy repeats, requiring copy number analysis in addition to standard sequencing for comprehensive “tma diagnosis”.
Screening for FH autoantibodies, particularly those targeting the C-terminus, is important for “tma diagnosis” of acquired complement-mediated aHUS. While FI autoantibodies exist, their pathogenic role is less clear. Besides complement gene screening, “tma diagnosis” should also include screening for other genetic TMAs like DGKE and MMACHC.
3.5. Genetic Complement-Mediated aHUS and Diagnostic Subtypes
Genetic mutations and autoantibodies are the main causes of complement-mediated aHUS. FH mutations are the most common, accounting for ~25% of cases and often affecting the C-terminus, impairing cell surface complement regulation. Loss-of-function mutations in Factor I and CD46 and activating mutations in C3 and complement factor B are also implicated in “tma diagnosis” of genetic subtypes of aHUS.
3.6. Predisposition, Penetrance, and Triggers in aHUS Diagnosis
Genetic variants associated with complement-mediated aHUS are predisposing rather than causative, with low penetrance. Disease development can occur throughout life, with penetrance increasing with multiple complement mutations. Single nucleotide polymorphisms (SNPs) can also modify penetrance. Environmental triggers are usually necessary to initiate aHUS in genetically predisposed individuals, highlighting the multifactorial nature of “tma diagnosis” in aHUS.
Infections, such as gastroenteritis and respiratory infections, are common triggers, implicated in about 50% of complement-mediated aHUS cases. Other triggers include pregnancy and certain drugs, which must be considered in “tma diagnosis” and patient history.
3.7. Acquired Complement-Mediated aHUS and Diagnostic Markers
FH antibody-mediated aHUS typically occurs in childhood and often follows gastrointestinal symptoms. It’s associated with homozygous deletion of CFHR3 and CFHR1, though the exact mechanism is unclear. FH autoantibody testing is crucial in “tma diagnosis” of this acquired form. While FI antibodies are found in some aHUS cases, their clinical significance is less established.
3.8. Treatment Strategies Following Complement-Mediated aHUS Diagnosis
Before complement inhibitors, plasma exchange (PE) was the primary treatment for complement-mediated aHUS post “tma diagnosis”. PE aimed to remove FH antibodies and overactive complement components and replace deficient regulatory proteins. PE is often initiated in adults with TMA while awaiting ADAMTS13 results to rule out TTP. In children, where TTP is rarer and PE has more risks, complement inhibitors are often first-line post “tma diagnosis”. PE remains the only option in regions with limited access to complement inhibitors due to cost.
Historically, PE alone had poor outcomes, with high rates of end-stage renal failure (ESRF) or death within a few years. Prognosis varied depending on the specific genetic mutation, with CD46 mutations generally having better outcomes and CFH mutations worse outcomes. Renal transplantation also had high recurrence rates (60%–70%) in aHUS, typically within the first year, except for patients with isolated CD46 mutations, emphasizing the importance of genetic “tma diagnosis” in predicting prognosis and recurrence risk.
3.8.1. Eculizumab Therapy Post-aHUS Diagnosis
Eculizumab revolutionized aHUS treatment post “tma diagnosis”. It’s a monoclonal antibody against complement C5, preventing its cleavage into C5a and C5b, thus blocking the terminal complement pathway. Eculizumab has dramatically improved outcomes and is considered safe in pregnancy.
Patients on eculizumab are at increased risk of encapsulated bacterial infections, especially Neisseria meningitides, requiring vaccination and prophylactic antibiotics. This highlights the importance of managing infectious risks associated with complement inhibition post “tma diagnosis” and treatment initiation.
3.8.2. Eculizumab Nonresponse and Differential Diagnosis
Eculizumab nonresponse occurs in some TMA cases. C5 polymorphisms (pR885H; pR885C) can prevent eculizumab binding, rendering it ineffective. In such cases, PE is recommended. Nonresponse can also occur in TMAs caused by non-complement gene mutations, such as MMACHC, DGKE, and INF2, emphasizing the need for comprehensive “tma diagnosis” beyond complement-mediated aHUS.
3.8.3. Ravulizumab: A Long-Acting Complement Inhibitor Post-Diagnosis
Ravulizumab, another C5 inhibitor, was developed as a longer-acting alternative to eculizumab. Engineered from eculizumab, it targets the same C5 epitope but has an extended half-life (~52 days vs. ~11 days for eculizumab), allowing for 8-week dosing intervals compared to eculizumab’s 2-week intervals.
Direct comparisons between ravulizumab and eculizumab in aHUS are lacking, as studies have been single-arm. Some concerns arose from the adult ravulizumab trial showing numerically more deaths and fewer patients discontinuing dialysis compared to historical eculizumab trials. However, low mutation/FH autoantibody rates in the ravulizumab trial suggest that many enrolled patients may not have had complement-mediated aHUS, potentially skewing results. Data suggest deaths in these trials were not complement-mediated aHUS-related (e.g., sepsis). The pediatric ravulizumab study showed more promising results, with higher mutation/FH autoantibody rates and 94.4% of patients achieving complete TMA response by 50 weeks, reinforcing the efficacy of C5 inhibition in appropriately diagnosed complement-mediated aHUS.
3.8.4. Disease-Driven vs. Continuous Eculizumab Therapy and Monitoring Post-Diagnosis
Eculizumab was initially approved for long-term aHUS treatment, but evidence supporting continuous therapy is limited. The incomplete penetrance of complement gene mutations and the historical success of withdrawing PE in some aHUS patients suggest continuous therapy may not always be necessary post “tma diagnosis”. Eculizumab has altered the natural history of aHUS, potentially preventing ESRF in patients who would have progressed despite PE, making relapse risk after withdrawal a key consideration.
Case reports and series have explored eculizumab withdrawal post “tma diagnosis”. Relapses occur in a proportion of patients after withdrawal, but prompt eculizumab re-initiation typically controls TMA and restores renal function. These reports suggest an intermittent, disease-driven eculizumab regimen may be feasible in select patients, although selection bias and non-prospective nature of these reports limit definitive conclusions. Relapses are more common in patients with pathogenic mutations, but not all mutation carriers relapse.
The STOPECU study, the first of three prospective trials on eculizumab withdrawal, suggests that withdrawal is possible in some aHUS patients post “tma diagnosis”. Relapse in STOPECU was primarily seen in patients with complement gene mutations. Female gender and elevated sC5b9 levels at withdrawal were associated with increased relapse risk. Most relapsing patients responded well to eculizumab re-treatment, but some had worse renal outcomes. Ongoing trials like SETSaHUS (UK) and CUREiHUS (Netherlands) will further refine eculizumab withdrawal strategies. A personalized approach to cessation, guided by mutation type, residual renal function, and transplant status, is likely to emerge. Optimal monitoring strategies for intermittent therapy, using urinalysis, blood pressure, and blood tests, are still being defined, emphasizing the evolving landscape of aHUS management post “tma diagnosis”.
4. Other Genetic TMAs: Diagnostic Considerations
4.1. Diacylglycerol Kinase Epsilon (DGKE) TMA: Diagnosis and Management
DGKE-related TMA is a rare aHUS variant (0.009/million/year in the UK) typically presenting in children under 2 years. First described in 2013, recessive DGKE mutations cause either aHUS or mesangioproliferative glomerulonephritis. Clinically, DGKE-TMA resembles complement-mediated aHUS, but with significant proteinuria. Viral triggers or prodromal diarrhea are sometimes present, complicating “tma diagnosis” differentiation.
While the exact mechanism is still under study, DGKE mutations disrupt VEGFR2 signaling and PGE2-mediated endothelial activation, leading to a prothrombotic state. No consistently effective treatment beyond supportive care exists. Relapses are common in early childhood. Many patients progress to ESRF but do not typically experience recurrence post-transplant, which is relevant for long-term management post “tma diagnosis”.
4.2. Cobalamin C aHUS: Diagnostic Clues and Treatment
Cobalamin C (vitamin B12) deficiency, due to recessive MMACHC mutations, is a metabolic disorder primarily affecting children. Elevated plasma homocysteine and urine/plasma methylmalonic acid levels are diagnostic markers. MMACHC deficiency impairs cobalamin conversion to its active forms, leading to homocysteine and methylmalonyl-CoA accumulation.
Cobalamin C deficiency usually presents with neurological, cardiac, ophthalmic, and developmental issues. A subset develops TMA, possibly due to homocysteine-induced endothelial damage. Treatment involves hydroxycobalamin and betaine. Complement inhibitors are ineffective, highlighting the importance of accurate “tma diagnosis” to guide appropriate therapy.
4.3. Infection-Associated TMAs: Diagnostic Differentiation
4.3.1. Shiga Toxin-Producing Escherichia coli (STEC)-HUS: Diagnosis and Management
STEC-HUS is the most common TMA, especially in children under 5, accounting for 90% of pediatric HUS cases. It follows infection with STEC, often from undercooked meat, unpasteurized dairy, or animal contact. Patients develop bloody diarrhea, abdominal pain, and vomiting about 3 days post-exposure. 10%–15% progress to HUS 7–10 days later.
STEC attaches to intestinal epithelium and produces Shiga toxin, which enters the bloodstream and binds to Gb3 receptors in kidneys, brain, and gut, causing cell death by inhibiting protein synthesis. Adults are less affected, possibly due to pre-existing anti-Shiga antibodies or lower Gb3 receptor expression.
Differentiating STEC-HUS from complement-mediated aHUS can be initially challenging, as ~30% of complement-mediated aHUS patients have diarrhea, and ~5% of STEC-HUS patients lack diarrhea. Fecal culture for E. coli O157:H7 and PCR for STEC genes are crucial for “tma diagnosis” in all HUS cases.
STEC-HUS is typically self-limiting, resolving within weeks with supportive care (fluids, dialysis, transfusions). However, ~30% develop long-term renal complications. No specific treatments exist. Complement activation occurs in STEC-HUS, but complement inhibitors are of uncertain benefit due to limited controlled data. Trials on eculizumab in STEC-HUS are ongoing (ECUSTEC ISRCTN89553116, NCT02205541).
4.3.2. Shigella dysenteriae Type 1 HUS: Diagnosis and Prognosis
Shigella dysenteriae type 1 also produces Shiga toxin and causes HUS, particularly in developing countries. Clinical features resemble STEC-HUS, but diarrhea may be initially watery then bloody/mucoid, and fever is common. HUS develops ~7 days after diarrhea resolves. While <10% of infected children develop TMA, mortality is high, and survivors are more prone to chronic kidney disease. “tma diagnosis” in this context is critical for supportive care and managing complications.
4.3.3. Pneumococcal HUS (pHUS): Diagnosis and Management
Invasive Streptococcus pneumoniae infection can cause TMA. Most cases present with pneumonia, but meningitis or ear/sinus infections also occur. pHUS patients are severely ill, often needing intensive care. Dialysis is needed in 75% of children, and ESRD develops in a third.
pHUS pathogenesis involves S. pneumoniae neuraminidase, which cleaves sialic acid from endothelial cells, platelets, and erythrocytes, exposing the Thomsen–Freidenreich (T) antigen. Pre-formed IgM antibodies bind T antigen, causing platelet aggregation, endothelial damage, and TMA. Coombs test is positive in pHUS, unlike other TMAs. Reduced FH binding due to sialic acid cleavage may also impair complement regulation and contribute to endothelial damage.
Treatment is supportive and targets the underlying infection. PE is controversial due to concerns about worsening disease by infusing anti-T IgM. Case studies using washed/plasma-reduced components like albumin with PE show promise, possibly by removing anti-T IgM and neuraminidase. Mortality remains high (~10%), emphasizing the severity and diagnostic challenges of pHUS.
4.3.4. Human Immunodeficiency Virus (HIV)-Associated TMA: Diagnosis and Treatment
HIV-associated TMA, likely due to endothelial damage, has become less common since highly active antiretroviral therapy (HAART), now ~0.3% incidence in HIV patients. Treatment is supportive and involves optimizing antiretroviral therapy. “tma diagnosis” in HIV patients requires considering TMA in the differential.
4.3.5. COVID-19 Infection and TMA: Diagnosis and Management
Severe acute respiratory syndrome-coronavirus-2 (SARS-CoV-2) can trigger complement-mediated aHUS. Marked complement activation and endothelial injury are seen in severe COVID-19. In individuals with latent complement dysregulation, COVID-19 can provoke aHUS. C5 inhibition is the logical treatment in COVID-19-induced complement-mediated aHUS, highlighting the importance of recognizing this TMA subtype in the context of severe infection.
COVID-19 can also cause broader thrombotic complications and AKI, even with anticoagulation. While C5 inhibition was proposed for these, a ravulizumab trial in COVID-19 was stopped due to lack of efficacy in preventing broader thrombotic events, suggesting targeted complement inhibition may be specific to aHUS but not general COVID-19 thrombotic complications.
4.3.6. Other Infections as TMA Triggers: Diagnostic Awareness
Many viral, bacterial, and parasitic infections have been linked to complement-mediated aHUS triggers. One study found infectious triggers in 73% of complement-mediated aHUS cases, emphasizing the broad range of infections to consider in “tma diagnosis” of aHUS, particularly in the context of TMA.
4.4. Pregnancy-Associated TMA: Diagnostic Challenges and Management
Pregnancy and postpartum periods are high-risk for both TTP and complement-mediated aHUS. Pregnancy-related TTP typically occurs in the second and third trimesters. Pregnant women account for up to a third of adult TTP cases, with ~25% being cTTP (vs. 5% in non-pregnancy-related adult TTP). Increased VWF production in pregnancy, leading to increased ADAMTS13 consumption, is thought to be the underlying cause, especially in women with pre-existing ADAMTS13 deficiency. Maternal and fetal mortality risks are high in pregnancy-related TTP, necessitating urgent PE. The PLASMIC score is not validated in pregnancy, making “tma diagnosis” rely more on clinical suspicion and ADAMTS13 testing.
Caplacizumab use in pregnancy is limited to case reports, and safety is unknown. Rituximab use in pregnancy is also cautious due to reported fetal abnormalities. Relapse risk in subsequent pregnancies is high. cTTP pregnancies typically relapse without prophylactic FFP infusions. aTTP relapse rate in pregnancy is ~50%. Pre-conception optimization of ADAMTS13 activity and antibody levels is recommended to prevent relapse. Rituximab can increase ADAMTS13 activity but conception should be delayed at least 6 months post-therapy.
Pregnancy-associated aHUS, unlike TTP, often occurs postpartum. Studies show ≥50% of pregnancy-related HUS cases have complement mutations, similar to general aHUS populations. Pregnancy is considered an aHUS trigger, and eculizumab is effective and safe in pregnancy-associated aHUS.
Pre-eclampsia, eclampsia, and HELLP syndrome are differential diagnoses for TTP and aHUS in pregnancy. Pre-eclampsia is characterized by hypertension and proteinuria. HELLP is a severe variant with glomerular endotheliosis. Pathogenesis involves endothelial dysfunction, possibly due to increased soluble vascular endothelial growth factor receptor (sFlt-1) and other antiangiogenic factors. While 8%–10% of pre-eclampsia/HELLP patients have complement gene variants, most are not pathogenic. Mouse models and elevated complement levels in pre-eclampsia suggest a complement role, but pre-eclampsia has occurred in women on eculizumab for other conditions, suggesting the terminal complement pathway may not be essential for pre-eclampsia pathogenesis.
Diagnostic tests for pregnancy-related TMA include blood pressure, proteinuria, liver enzymes (elevated in HELLP), and sFlt-1/PlGF ratios to assess pre-eclampsia risk. Ratios indicative of pre-eclampsia/HELLP are >110 after 34 weeks gestation and >85 before 34 weeks. Rapid and accurate “tma diagnosis” and management are critical in pregnancy-related TMAs due to maternal and fetal risks. International guidelines for TMA assessment and management in pregnancy exist, emphasizing the complexity and urgency of “tma diagnosis” in this setting.
4.5. Drug-Induced TMA: Diagnosis and Causality
Many drugs are associated with TMA, but causality is established for only a few. Two main mechanisms are immune-mediated and direct toxicity. Quinine can induce autoantibodies to platelet glycoproteins, causing immune-mediated TMA. Drugs with proven direct toxicity include interferon β and bevacizumab. Chemotherapy agents (e.g., gemcitabine) and immunosuppressants (e.g., calcineurin inhibitors) may also be implicated. Treatment involves stopping the suspected drug and supportive care. Ticlopidine can cause TTP with ADAMTS13 antibodies, requiring TTP-specific treatment and ticlopidine withdrawal. “tma diagnosis” in drug-induced cases hinges on identifying and discontinuing the causative agent.
4.6. De Novo TMA Post-Solid Organ Transplant: Diagnosis and Management
De novo TMAs occur post-solid organ transplant. Pathogenesis is multifactorial, involving antibody-mediated rejection, ischemia-reperfusion injury, calcineurin inhibitors, and infections, all leading to endothelial damage. Underlying complement mutations are found in ~29% of renal transplant recipients who develop TMA. Treatment involves adjusting immunosuppression, treating infections, and considering complement inhibitors if complement-mediated aHUS is suspected. “tma diagnosis” in transplant settings requires considering these multiple factors.
4.7. Bone Marrow Transplant TMA: Diagnosis and Treatment
TMA post-bone marrow transplant is a serious complication with high mortality. Etiology is complex, possibly involving vascular graft-versus-host disease (GVHD) due to association with severe GVHD. Some evidence of complement activation and rare aHUS-associated gene mutations exist. Eculizumab has been used, but prospective trial data is awaited. “tma diagnosis” in bone marrow transplant recipients is challenging and requires multidisciplinary approach.
4.8. Malignancy-Associated TMA: Diagnosis and Prognosis
Malignancy-related TMA can be due to the malignancy itself or chemotherapy. Differentiating between the two is often difficult. Tumor emboli in microvasculature may directly shear erythrocytes. Treatment is supportive and includes stopping chemotherapy, but prognosis is poor due to the underlying malignancy. “tma diagnosis” in cancer patients should consider both direct and treatment-related causes.
4.9. Autoimmune TMA: Diagnosis and Specific Conditions
TMAs are recognized complications in systemic lupus erythematosus (SLE), scleroderma renal crisis (SRC), and catastrophic antiphospholipid syndrome (CAPS). Mechanisms are unclear.
In SLE, complement’s classical pathway is a key disease driver, with alternative pathway involvement. However, no clear correlation between complement activity and SLE-TMA exists. Case reports suggest eculizumab response in SLE-TMA, but larger series have not confirmed this. Standard immunosuppression remains the primary treatment for SLE-TMA.
CAPS has renal TMA in ~30% of cases, with high overall mortality (~36%). CAPS triple therapy (glucocorticoids, anticoagulants, and PE/IVIG) improves mortality compared to other regimens. Complement activation is evident in CAPS, and case reports suggest eculizumab benefit.
Evidence for complement’s role in SRC is limited. TMA occurs in 45%–50% of SRC cases. ACE inhibitors have significantly reduced SRC mortality. “tma diagnosis” in autoimmune conditions requires considering the underlying autoimmune disease and its specific TMA manifestations.
4.10. TMA Associated with Glomerular Disease: Diagnostic Overlap
Histopathological TMA features are seen in various glomerular diseases without typical MAHA or thrombocytopenia, such as IgA nephropathy, ANCA-associated vasculitis, membranous nephropathy, focal segmental glomerulosclerosis, and C3 glomerulopathy (C3G)/mesangioproliferative glomerulonephritis (MPGN).
C3G/MPGN are complement-mediated but differ from aHUS in pathogenesis. C3G involves fluid-phase complement activation, while aHUS is characterized by solid-phase cell surface activation. Concurrent and sequential C3G and TMA cases have been reported, highlighting diagnostic complexity and overlap.
4.11. Hypertensive TMA: Diagnostic Mimicry and Differentiation
Severe hypertension can cause pathological TMA and clinical TMA features (AKI, MAHA, thrombocytopenia), mimicking complement-mediated aHUS. A retrospective study found that many patients initially diagnosed with hypertensive TMA who progressed to ESRF despite blood pressure control actually had complement-mediated aHUS based on genetic analysis. Failure to control MAHA and thrombocytopenia with blood pressure management should prompt consideration of PE or eculizumab while awaiting complement evaluation. Hypertensive TMA often progresses to ESRF, and genetic analysis should be done before renal transplant listing to differentiate from aHUS. “tma diagnosis” must carefully distinguish hypertensive TMA from aHUS, particularly in cases with severe hypertension.
4.12. Summary: Advancements in TMA Diagnosis and Treatment
TMAs present with diverse clinical manifestations. Recent research has greatly advanced our understanding of TMA pathogenesis and led to targeted therapies, significantly improving patient outcomes. Accurate and timely “tma diagnosis” remains crucial for effective management and leveraging these therapeutic advances across the spectrum of TMA subtypes.
CONFLICT OF INTEREST
David Kavanagh has received consultancy income from Gyroscope Therapeutics, Silence Therapeutics, Alexion Pharmaceuticals, Novartis, Apellis, and Sarepta.
Thompson GL, Kavanagh D. Diagnosis and treatment of thrombotic microangiopathy. Int J Lab Hematol. 2022;44(S1):101‐113. doi: 10.1111/ijlh.13954
DATA AVAILABILITY STATEMENT
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
REFERENCES
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.