Transthyretin amyloid cardiomyopathy (ATTR-CM) represents a serious and frequently underdiagnosed cause of heart failure that, crucially, is now treatable. The challenges in diagnosis often lead to delays or misdiagnosis, significantly impacting patient outcomes. With the advent of effective therapies like tafamidis, timely and accurate diagnosis is more critical than ever. Modern, cost-effective imaging techniques, such as longitudinal strain imaging, cardiac magnetic resonance (CMR) T1 mapping, and cardiac scintigraphy, have revolutionized the diagnostic approach to ATTR-CM, reducing the need for invasive tissue biopsies. This article provides an updated review of current diagnostic tools, empowering clinicians to confidently diagnose ATTR-CM.
Understanding TTR Amyloidosis: Subtypes and Pathogenesis
Transthyretin (TTR) amyloid cardiomyopathy (ATTR-CM) arises from the deposition of misfolded transthyretin protein within the heart muscle. Unlike immunoglobulin light chain (AL) amyloidosis, where misfolded light chains from plasma cells are the culprit [1], ATTR-CM involves TTR, a protein primarily produced in the liver. This misfolding process results in the formation of amyloid fibrils, characterized by a cross-beta-sheet structure, which are toxic to various tissues.
Diagnosing ATTR-CM can be complex because its clinical presentation overlaps with many other cardiac conditions. Furthermore, differentiating ATTR-CM from AL amyloidosis is crucial for appropriate management, yet often poses a challenge for clinicians. Misdiagnosis and subsequent treatment for the incorrect condition are alarmingly common in ATTR-CM patients [2]. Therefore, a strong clinical suspicion and the strategic use of diagnostic tools are paramount. This review will detail the essential diagnostic modalities for accurate Ttr Amyloidosis Diagnosis.
Prevalence and Subtypes of ATTR-CM
The precise prevalence of ATTR-CM remains elusive due to underdiagnosis. Wild-type ATTR-CM (ATTR-CMwt) is the more prevalent form, with incidence increasing significantly with age, as demonstrated in autopsy studies [3]. Bone scintigraphy studies in older adults hospitalized with heart failure with preserved ejection fraction (HFpEF) revealed ATTR-CM in 13% of cases, with all diagnoses being ATTR-CMwt by the age of 86 [4]. Hereditary ATTR-CM (ATTR-CMh) shares a similar clinical presentation to ATTR-CMwt, typically manifesting as late-onset restrictive cardiomyopathy, with an average age of presentation around 69 years [5].
ATTR-CM is genetically categorized based on the TTR gene. ATTR-CMwt occurs without any identifiable TTR gene mutation, while ATTR-CMh is characterized by a specific single amino acid mutation within the TTR gene. Notably, genetic studies indicate that ATTR-CMh is present in approximately 3.5% of African Americans, and this allele frequency rises to 10% in African Americans over 65 with congestive heart failure [6]. Table 1 summarizes the key distinctions between ATTR-CMwt and ATTR-CMh.
Table 1. Similarities and differences between wild-type and hereditary amyloid transthyretin cardiomyopathy
| Feature | ATTR-CMwt | ATTR-CMh |
|---|---|---|
| Age of onset | Typically > 60 years | Variable depending on mutation (30–80 years) |
| Genotype | Normal | Abnormal, nucleotide mutations present |
| Survival | Approximately 3.5 years | Variable depending on genetic mutation |
| Patient demographics | Male predominance, increased prevalence with age, African American | Mutations endemic to certain locations (Ireland, Japan, Sub-Saharan Africa) |
TTR, in its native state, is a tetramer composed of four beta-sheet-rich monomers, secreted by the liver, choroid plexus, and retinal epithelial cells. It functions as a transport protein for thyroxine and holo-retinol binding protein [7]. In ATTR-CMh, a single amino acid mutation in the 127 amino acid sequence of TTR disrupts its stability, leading to misfolding and aggregation [8]. This misfolded TTR infiltrates various tissues, causing ATTR amyloidosis [9]. In the heart, aggregated and misfolded TTR creates a rigid, space-occupying infiltrate, resulting in myocardial restriction and dysfunction [10].
Clinical Manifestations: Cardiac and Extracardiac Clues for TTR Amyloidosis Diagnosis
Cardiac Involvement and Differential Diagnoses
The typical ATTR-CM patient is an older individual presenting with heart failure with preserved ejection fraction (HFpEF). Their clinical history often reveals progressively worsening heart failure unresponsive to standard treatments. Echocardiographic findings, as detailed below, commonly show increased left ventricular wall thickness, and despite therapy, their condition deteriorates. Hemodynamically, they often exhibit a restrictive physiology. Notably, these patients may not tolerate beta-blockers and may experience spontaneous improvement in hypertension, even without medication or lifestyle changes [10]. The average age at diagnosis for ATTR-CMwt is 74 years, with a higher prevalence in males [11], sometimes referred to as “senile” amyloidosis in this demographic.
TTR amyloidosis diagnosis can be challenging due to its resemblance to other cardiac conditions, primarily aortic stenosis, hypertrophic cardiomyopathy, and AL amyloidosis. Other conditions that can mimic ATTR-CM include dilated cardiomyopathies, toxic cardiomyopathies, peripartum cardiomyopathy, cardiac sarcoidosis, and stress-induced cardiomyopathy. This overlap often leads to underdiagnosis of ATTR-CM in patients initially diagnosed with these conditions.
Studies have indicated a significant prevalence of ATTR-CM in patients with severe aortic stenosis undergoing surgical valve replacement [12]. In patients with low-flow, low-gradient severe aortic stenosis, the persistence of restrictive physiology and limited improvement post-valve replacement may be attributed to coexisting ATTR-CM. These patients have demonstrated poorer outcomes despite valve replacement and hemodynamic improvements [13]. Ongoing research into long-term outcomes post-transcatheter aortic valve replacement will further elucidate the role of ATTR-CM in these patients.
Asymmetric septal hypertrophy, mimicking hypertrophic cardiomyopathy, has been observed in up to 25% of ATTR-CMwt patients [2]. Incidental ATTR-CM diagnoses have been made histologically in patients undergoing septal myectomy for left ventricular outflow tract (LVOT) obstruction [14]. Clinicians managing hypertrophic cardiomyopathy patients should consider ATTR-CM as a differential diagnosis, and histopathological examination should be performed on septal myectomy specimens to rule out ATTR-CM.
Extracardiac Involvement: Early Warning Signs
Extracardiac manifestations of ATTR can serve as crucial early indicators, enabling clinicians to consider TTR amyloidosis diagnosis even before cardiac symptoms become prominent. These manifestations include involvement of the central nervous system, kidneys (nephrotic syndrome), eyes, gastrointestinal system, autonomic and peripheral nervous systems. However, the most frequently observed extracardiac findings in ATTR are bilateral carpal tunnel syndrome (CTS), spinal stenosis, and spontaneous biceps tendon rupture [15, 16]. While ATTR-CMwt and ATTR-CMh generally share similar extracardiac manifestations, genetic variations in ATTR-CMh can lead to slight differences. For instance, the VAL122lle (pV1421) mutation typically does not cause polyneuropathy [17], whereas the Thr60AIa (pT80A) mutation, prevalent in Ireland, is associated with a high incidence of carpal tunnel syndrome [18].
Amyloid fibril infiltration in soft tissues can cause nerve entrapment, with carpal tunnel syndrome being the most common. Remarkably, CTS symptoms can precede cardiac manifestations of ATTR-CM by up to 10 years [19]. Approximately 50% of ATTR-CMwt patients present with carpal tunnel syndrome [20]. The presence of carpal tunnel syndrome, particularly in patients with heart failure symptoms, should raise suspicion for ATTR-CM, facilitating earlier diagnosis. Furthermore, pathological examination of tenosynovial tissue from carpal tunnel release surgery can reveal amyloid deposits, as demonstrated in previous studies [21].
Similar to CTS, ATTR deposition in musculoskeletal tissues can lead to spinal canal narrowing due to ligamentum flavum compression. Studies have shown ATTR deposition in over 45% of older patients undergoing surgery for spinal stenosis [22]. Additionally, ATTR deposits were found in 33% of patients with spontaneous distal biceps tendon rupture [15]. Amyloid deposits have also been identified in tissue samples from patients undergoing rotator cuff repairs [23] and total knee and hip arthroplasties [24]. These musculoskeletal manifestations can serve as important clinical clues for screening patients at risk of ATTR-CM.
Diagnostic Tools for TTR Amyloidosis Diagnosis
Serology and Biomarkers
Unlike AL amyloidosis, which has circulating biomarkers like light chains, ATTR-CM lacks specific serological biomarkers currently in routine clinical use. Novel tests for endogenous TTR ligand retinol binding protein 4 are being investigated as potential future biomarkers [25]. Traditional cardiac biomarkers, such as troponin and pro-B-type natriuretic peptide (BNP), are consistently elevated in ATTR-CM and are incorporated into staging and prognostic assessments [26]. Serum troponin levels may be persistently elevated even in the absence of overt cardiomyopathy, and BNP levels are often disproportionately high relative to the clinical severity of heart failure [27]. While not specific for TTR amyloidosis diagnosis, these biomarkers contribute to risk stratification and monitoring.
Electrocardiogram (ECG)
An ECG can provide initial clues for TTR amyloidosis diagnosis. Patients with increased left ventricular wall thickness on echocardiography who present with low-voltage ECG should be further evaluated for ATTR-CM. Typically, conditions like hypertrophic cardiomyopathy and hypertensive heart disease, which also cause increased wall thickness, are associated with ECG criteria for left ventricular hypertrophy, not low voltage. However, low voltage is not universally present in ATTR-CM, with studies showing it in only up to 40% of patients [28].
Atrioventricular (AV) block is another significant ECG finding in ATTR-CM. Amyloid infiltration of the sinus and AV nodes can disrupt the electrical conduction system, often necessitating pacemaker implantation. AV block is observed in up to 22% of cardiac amyloidosis cases [29].
Imaging Modalities: Non-invasive TTR Amyloidosis Diagnosis
Imaging is central to non-invasive TTR amyloidosis diagnosis. Transthoracic echocardiography, cardiovascular magnetic resonance (CMR), and cardiac scintigraphy are the three primary imaging modalities used, each offering unique diagnostic insights.
Transthoracic Echocardiography
Echocardiography provides several findings suggestive of cardiac amyloidosis. Symmetrical thickening of the left ventricular (LV) walls, often misdiagnosed as LV hypertrophy, is a key feature. In contrast to true LV hypertrophy from myocyte enlargement, amyloidosis involves extracellular amyloid protein deposition with normal-sized myocytes. The thickened myocardium often exhibits a characteristic “sparkling” or “speckled appearance” due to amyloid infiltration. Pleural and pericardial effusions, though often small, may also be present. Diastolic dysfunction with a restrictive filling pattern and bi-atrial enlargement are common but typically seen in later stages [30]. However, these echocardiographic findings are not entirely specific, as increased LV wall thickness and speckled patterns can also occur in conditions like end-stage renal disease and glycogen storage diseases.
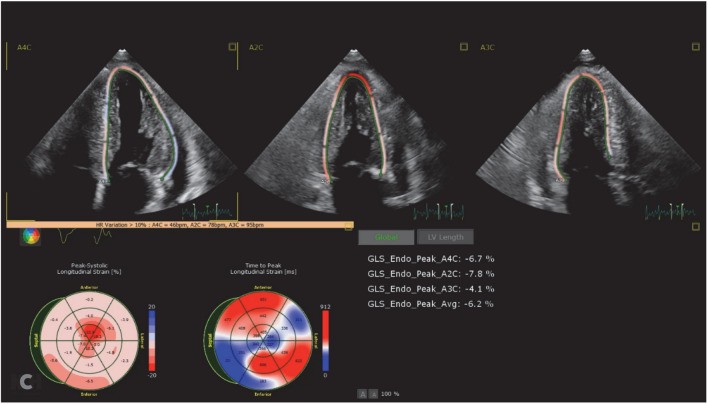
Advanced echocardiographic techniques, particularly longitudinal systolic strain imaging, have significantly enhanced diagnostic accuracy. Longitudinal strain is reduced in ATTR-CM, characteristically showing preserved strain in the apical region of the left ventricle while the mid and basal regions are more severely affected. This creates a distinctive “cherry on top” pattern on longitudinal strain image maps (Fig. 1). An abnormal apical-to-mid or basal strain ratio is highly suggestive of cardiac amyloidosis and aids in differentiating it from other conditions [31]. While highly suggestive of cardiac amyloidosis, strain imaging alone cannot differentiate between ATTR-CM and AL amyloidosis, as both can exhibit similar patterns.
Fig. 1. Transthoracic echocardiography demonstrating a classic cardiac amyloid longitudinal strain pattern of the “cherry on top”
Cardiovascular Magnetic Resonance (CMR) Imaging
CMR has become increasingly valuable in evaluating various cardiac pathologies, including infiltrative cardiomyopathies like ATTR-CM. Gadolinium contrast enhancement is crucial for detecting amyloid infiltration. In amyloidosis, gadolinium is retained within the myocardium, resulting in an inability to suppress the myocardial signal, or diffuse subendocardial or transmural late gadolinium enhancement (LGE). This pattern is highly sensitive and specific for amyloidosis [32]. In contrast, other conditions like hypertensive heart disease typically show washout of gadolinium from the myocardium, without this persistent enhancement (Fig. 2).
Fig. 2. CMR images demonstrating late gadolinium enhancement in cardiac amyloidosis.
a Four-chamber phase-sensitive inversion recovery sequences (inversion time 240 ms) 10-min post gadolinium demonstrated late gadolinium enhancement in all four chambers, inability to null the myocardium and transmural involvement. b Short-axis phase-sensitive inversion recovery sequence (inversion time 240 ms) 10-min post gadolinium demonstrated late gadolinium enhancement in all four chambers, inability to null the myocardium and transmural involvement.
Figure images courtesy of Dr Mohammad Al-Ani, Division of Cardiovascular Medicine, University of Florida, Gainesville, Florida
Advanced CMR techniques like T1 mapping further enhance diagnostic sensitivity and provide quantitative measures of myocardial amyloid infiltration. T1 mapping quantifies the relaxation times of myocardial tissue, and parameters such as extracellular volume fraction (ECV) are significantly elevated in cardiac amyloidosis compared to other myocardial diseases [33].
While CMR is highly effective in identifying infiltrative processes like cardiac amyloidosis, it cannot reliably differentiate between ATTR-CM and AL amyloidosis [34]. Therefore, additional modalities are needed for definitive TTR amyloidosis diagnosis and subtype identification.
Cardiac Scintigraphy with 99m Technetium Pyrophosphate (TC-99m PYP)
Cardiac scintigraphy using bone-avid radiotracers is currently the only imaging modality that can accurately differentiate between ATTR-CM and AL amyloidosis non-invasively.
Technetium-labeled radiotracers, including TC-99m-PYP, TC-99m-3,3-diphosphono-1,2-propanodicarboxylic acid, and TC-99m-hydroxymethylene diphosphonate, are used to detect ATTR-CM [10]. Myocardial uptake of these tracers is compared to rib uptake, with cardiac uptake graded visually. Cardiac uptake greater than rib uptake is considered positive. While the exact mechanism of radiotracer uptake is not fully understood, it is highly specific for ATTR-CM. When grade 2 or 3 cardiac uptake is observed in heart failure patients without monoclonal protein (ruling out AL amyloidosis) and with supportive echocardiographic or CMR findings, cardiac scintigraphy has demonstrated 100% specificity for ATTR-CM [36]. Thus, cardiac scintigraphy offers a powerful non-invasive method for TTR amyloidosis diagnosis.
Despite its high specificity for ATTR-CM, cardiac scintigraphy requires careful interpretation and exclusion of AL amyloidosis. A multidisciplinary approach, involving hematologic and nephrologic evaluations to rule out AL amyloidosis, remains essential. AL amyloid cardiomyopathy can also exhibit radiotracer uptake on cardiac scintigraphy. Standard serological tests for AL amyloidosis, such as serum and urine protein electrophoresis, can be challenging to interpret for non-specialists and may be unreliable. Furthermore, monoclonal gammopathy can coexist with ATTR-CM [37]. Therefore, a multidisciplinary evaluation, potentially including genetic testing and tissue biopsy in complex cases, is crucial for accurate diagnosis. Table 2 summarizes noninvasive diagnostic indicators for suspected ATTR-CM.
Table 2. Noninvasive diagnostic tests and indicators of suspected ATTR-CM
| Test | Salient features |
|---|---|
| Electrocardiogram | Discrepancy between left ventricular thickness and QRS voltage |
| Echocardiogram | Increased left ventricular wall thickness, Reduced longitudinal strain with apical sparing on strain imaging |
| Cardiac magnetic resonance imaging | Marked extracellular volume expansion, Abnormal nulling of myocardium on T1 mapping, Delayed gadolinium enhancement |
| Cardiac scintigraphy | Increased radiotracer uptake |
| Serology | Mild chronic cardiac biomarker elevation, Lack of biomarkers for amyloid light-chain amyloidosis |
| Salient clinical features | Heart failure with preserved ejection fraction, Intolerance to beta-blockers or ACE inhibitors, No longer hypertensive, Neuropathy, Carpal tunnel syndrome, Biceps tendon rupture, Lumbar spinal stenosis |
Genetic Testing in TTR Amyloidosis Diagnosis
Genetic testing is recommended for all patients diagnosed with ATTR-CM, regardless of age, due to the significant implications for family members. Currently, guidelines for surveillance in individuals carrying a TTR variant are still evolving [10]. Genetic testing helps differentiate between ATTR-CMwt and ATTR-CMh, provides information about specific mutations in hereditary forms, and facilitates family screening and genetic counseling.
Endomyocardial Biopsy: The Role of Tissue Diagnosis
Tissue biopsy, with histopathology and immunohistochemistry, has been the traditional gold standard for diagnosing amyloidosis. Extracardiac tissue biopsies, such as abdominal fat pad aspirates, have variable sensitivity and may be unreliable [38]. Tissue samples from orthopedic procedures, like carpal tunnel release surgery, may be diagnostically useful but their reliability is still under investigation.
Endomyocardial biopsy, traditionally considered the gold standard, offers high sensitivity and specificity, especially with multiple myocardial samples and Congo Red staining for amyloid detection [39]. Advanced techniques like tandem mass spectrometry analysis can further refine the diagnosis and confirm ATTR-CM subtype.
However, with advancements in non-invasive imaging, the need for endomyocardial biopsy is decreasing, particularly given the inherent risks associated with the procedure. Endomyocardial biopsy remains valuable in cases where AL amyloidosis cannot be confidently excluded by non-invasive methods.
Disease Progression and Prognosis in TTR Amyloidosis
The clinical course of ATTR-CM varies depending on whether it is ATTR-CMh or ATTR-CMwt. Both subtypes typically progress to overt heart failure and conduction system disease [28]. ATTR-CM generally follows a slowly progressive course, in contrast to the more rapidly progressing and severe heart failure seen in AL amyloidosis [28]. ATTR-CMh can present primarily as cardiomyopathy or neuropathy, although mixed phenotypes can occur, especially in late-diagnosed cases [40]. The natural history of ATTR-CMh is further influenced by specific mutations, fibril type, and individual genetic factors. Cardiac involvement is a major determinant of prognosis. Median survival for ATTR-CMh with polyneuropathy is reported around 10 years, compared to approximately 3 years for ATTR-CMh primarily presenting as cardiomyopathy [40]. ATTR-CMwt typically has a more consistent disease progression, with a median survival of approximately 3.5 years from diagnosis [10]. Disease stage at diagnosis also significantly impacts prognosis. Staging systems, like the Mayo Clinic staging system for ATTR-CMwt, incorporate biomarkers such as troponin T and N-terminal pro-B type natriuretic peptide levels to predict survival [41]. Conduction system disease, manifesting as higher rates of permanent pacemakers and atrial arrhythmias like atrial fibrillation, is more prevalent in ATTR-CMwt [41].
Conclusions: Advancing TTR Amyloidosis Diagnosis
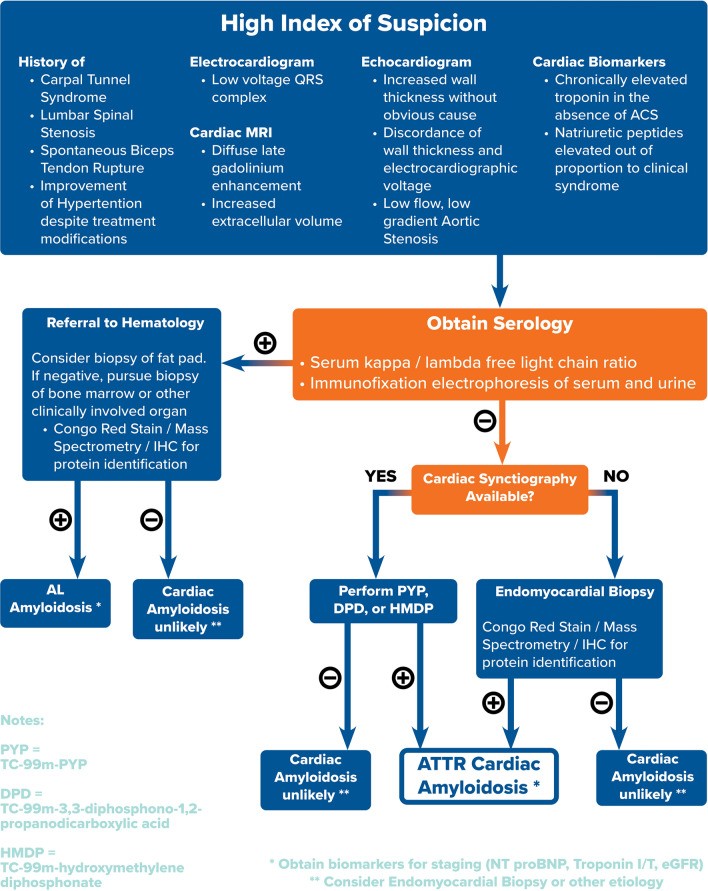
ATTR-CM is no longer an obscure and untreatable condition. Clinical cardiologists now have access to powerful non-invasive tools for screening and diagnosing ATTR-CM early, often years before significant cardiac damage occurs. Musculoskeletal and neuropathic symptoms can provide early clinical clues. Baseline ECG and echocardiography with longitudinal strain imaging can raise clinical suspicion. Cardiac scintigraphy offers the ability to differentiate ATTR-CM from other amyloid subtypes. Combined with serological testing to exclude AL amyloidosis and careful clinical evaluation, clinicians can confidently establish a non-invasive TTR amyloidosis diagnosis. Figure 3 outlines a proposed algorithmic approach to TTR amyloidosis diagnosis.
Fig. 3. Algorithmic approach to the diagnosis of ATTR-CM
Acknowledgements
Funding
No funding or sponsorship was received for this study or publication of this article.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Disclosures
Anthony A. Bavry is an Editorial Board member of the journal; Current affiliation is Department of Medicine, University of Texas Southwestern, Dallas, Texas. Adam S. Hafeez has nothing to disclose.
Compliance with Ethics Guidelines
This article does not contain any studies with human participants or animals performed by any of the authors.
Data Availability
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
Footnotes
Digital Features
To view digital features for this article go to https://doi.org/10.6084/m9.figshare.12032166.
References
[References will be listed here as in the original article]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
References
[1] … (References from original article to be included here in the final version)
[2] …
[3] …
[4] …
[5] …
[6] …
[7] …
[8] …
[9] …
[10] …
[11] …
[12] …
[13] …
[14] …
[15] …
[16] …
[17] …
[18] …
[19] …
[20] …
[21] …
[22] …
[23] …
[24] …
[25] …
[26] …
[27] …
[28] …
[29] …
[30] …
[31] …
[32] …
[33] …
[34] …
[35] …
[36] …
[37] …
[38] …
[39] …
[40] …
[41] …