Von Willebrand disease (VWD) stands out as the most prevalent inherited bleeding disorder, yet paradoxically, it is often the most challenging to accurately diagnose. This condition primarily manifests through mild mucosal bleeding, but can also present surgical bleeding complications and, in severe instances, joint bleeding. A significant clue in diagnosis is often a family history of VWD or unexplained bleeding tendencies. However, pinpointing VWD in the laboratory is not straightforward. It requires a series of specialized assays designed to evaluate both the quantity and function of von Willebrand factor (VWF), along with factor VIII activity. Currently, there isn’t a single, definitive diagnostic test to definitively confirm or rule out VWD. The landscape of VWD diagnosis is evolving, with newer VWF function assays becoming increasingly available and proving invaluable in refining laboratory diagnosis.
Keywords: Von Willebrand Factor Diagnosis, Von Willebrand disease, VWF testing, bleeding disorders, hemostasis
Understanding Von Willebrand Disease and the Role of Von Willebrand Factor
Von Willebrand disease (VWD) is a genetic bleeding disorder that arises from a deficiency in von Willebrand factor (VWF). It is typically characterized by mild mucosal bleeding. VWF is a crucial protein in the blood clotting process, playing multiple vital roles in hemostasis. One of its primary functions is to act as a bridge, linking platelets to the sites of blood vessel injury. It achieves this by binding to both collagen, a structural protein in blood vessel walls, and glycoprotein Ibα (GPIbα) on the surface of platelets. Specifically, VWF’s A1 domain binds to platelet GPIbα, while its A1 and A3 domains interact with vascular collagens [1, 2]. Beyond platelet interaction, VWF also plays a protective role for circulating factor VIII (FVIII). By binding to FVIII within its D’D3 domain, VWF prevents FVIII from premature degradation and ensures its availability in the coagulation cascade [3].
Patients with VWD most commonly experience mucosal bleeding. This can include nosebleeds (epistaxis), easy bruising, heavy menstrual bleeding (menorrhagia), gum bleeding, gastrointestinal (GI) bleeding, and bleeding following surgery, particularly procedures involving mucosal surfaces such as tonsillectomies or wisdom tooth extractions. In rarer and more severe forms, joint bleeding can also occur. Given its inherited nature, a family history of bleeding symptoms or diagnosed VWD is common, although the severity and type of symptoms can vary considerably among affected family members.
The underlying cause of VWD can be either a quantitative deficiency, meaning there isn’t enough VWF protein, or a qualitative defect, where the VWF protein is present but doesn’t function correctly. Quantitative defects are further categorized clinically into type 1 VWD, characterized by mild to moderate reductions in VWF, and type 3 VWD, the most severe form with virtually undetectable VWF levels. Qualitative defects fall under type 2 VWD, which is further subdivided based on the specific functional abnormality in the VWF protein. The current classification system divides type 2 VWD into four main subtypes [4]. Type 2A variants are characterized by the absence of high molecular weight multimers of VWF, which are crucial for effective platelet adhesion. Type 2B variants also lack these high molecular weight multimers. However, in type 2B, this is due to gain-of-function mutations in VWF, causing it to spontaneously bind to platelet GPIb. This leads to the clearance of VWF–platelet complexes from the circulation. Platelet-type VWD is a related disorder caused by a similar gain-of-function defect, but in platelet GPIb itself. Type 2M VWD encompasses variants with impaired platelet binding, despite having a normal multimer distribution. It also includes defects in collagen binding. Lastly, type 2N variants are defined by a specific defect in their ability to bind and protect factor VIII.
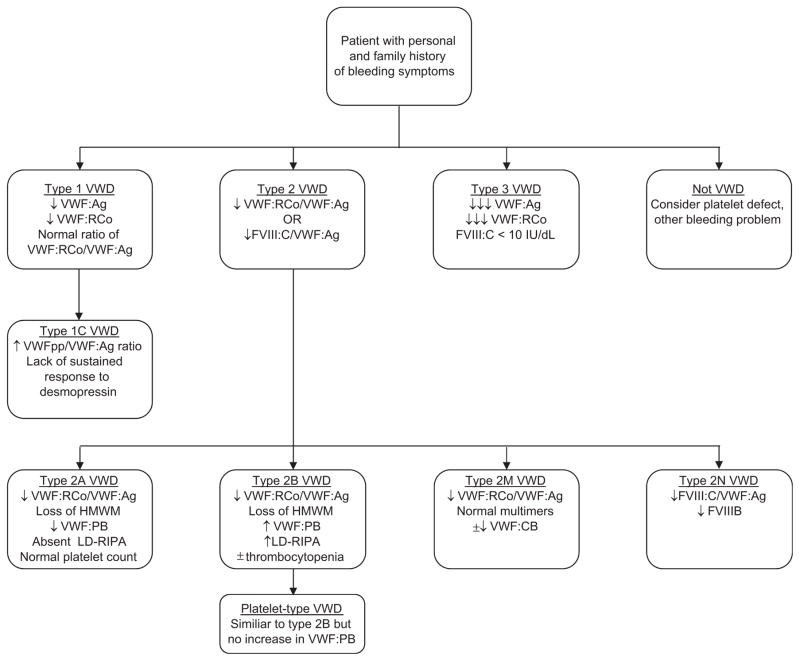
Initial laboratory evaluation for VWD requires a strategic combination of screening tests. No single test is sufficient to confirm the presence of fully functional VWF. Therefore, alongside assessing the amount of VWF protein, routine screening must include evaluations of VWF’s interactions with both platelets and factor VIII. A detailed overview of VWF testing is provided in Table 1, and a diagnostic algorithm for VWD is illustrated in Figure 1.
 Flow diagram for diagnosis of VWD
Flow diagram for diagnosis of VWD
Laboratory Diagnosis of Von Willebrand Disease: Screening Tests
Diagnosing Von Willebrand Disease involves a multi-step laboratory approach, beginning with screening tests designed to broadly assess VWF quantity and function. These initial tests help identify potential VWD cases and guide further, more specific confirmatory testing.
VWF Antigen (VWF:Ag)
The VWF antigen test (VWF:Ag) is a quantitative assay that measures the level of VWF protein present in plasma. This test is typically performed using either an enzyme-linked immunosorbent assay (ELISA) or an automated latex immunoassay (LIA) [5]. VWF:Ag assays are generally considered reliable and reproducible for determining the amount of VWF protein. However, a key limitation is that VWF:Ag only assesses the quantity of VWF and does not provide information about its functional capabilities. Consequently, VWD screening necessitates additional tests that specifically evaluate VWF function to detect any functional defects in the VWF protein.
VWF Ristocetin Cofactor Activity (VWF:RCo)
The VWF ristocetin cofactor activity assay (VWF:RCo) is the most commonly used functional assay to assess VWF’s ability to bind to platelet GPIbα. Ristocetin is an antibiotic that, in laboratory settings, promotes the binding of VWF to GPIbα, mimicking the interaction necessary for platelet adhesion at sites of vascular injury. The VWF:RCo assay measures how effectively VWF in a patient’s plasma can facilitate platelet aggregation in the presence of ristocetin.
While widely used, the VWF:RCo assay has limitations, including a relatively high coefficient of variation, which can affect its precision [6]. Furthermore, genetic variations within the VWF A1 domain, the region responsible for GPIbα binding, can influence VWF:RCo results without necessarily indicating a true functional defect in vivo [7]. To overcome these limitations, newer assays that directly evaluate VWF-GPIbα interactions independent of ristocetin have been developed [8, 9]. These assays hold promise for more accurately assessing VWF function related to platelet binding. While not yet commercially available in the US, a reliable assay of VWF–platelet interactions could potentially replace the VWF:RCo and might even eliminate the need for VWF:Ag in initial VWD screening. However, it’s important to note that such an approach would still miss type 2M VWD variants that primarily affect collagen binding.
Factor VIII Activity (FVIII:C)
Measurement of factor VIII activity (FVIII:C) is an integral part of the initial VWD diagnostic workup. This is because VWF acts as a carrier protein for FVIII in the circulation, protecting it from degradation and delivering it to sites of coagulation. Under normal physiological conditions, the ratio of FVIII:C to VWF:Ag is approximately 1. In type 2N VWD, where there is a specific defect in VWF’s ability to bind FVIII, this ratio will be decreased. In type 3 VWD, characterized by a near-complete absence of VWF, FVIII:C levels will also be significantly reduced due to the lack of VWF protection. Therefore, FVIII:C measurement provides valuable information about VWF function, particularly in relation to FVIII binding and protection.
VWF:RCo/VWF:Ag Ratio
Assessing the ratio of VWF:RCo to VWF:Ag is a crucial step in the VWD diagnostic process. A reduced VWF:RCo/VWF:Ag ratio is a key indicator that prompts further investigation for type 2A, type 2B, or type 2M VWD. These type 2 variants are characterized by qualitative defects in VWF function, leading to a disproportionate reduction in VWF activity (VWF:RCo) compared to the amount of VWF protein present (VWF:Ag). Generally, a VWF:RCo/VWF:Ag ratio below 0.6 is suggestive of a type 2 VWD variant [10]. However, it’s important to emphasize that while a decreased ratio is suggestive, confirmatory testing is always necessary to definitively distinguish between the different type 2 VWD subtypes and to confirm the diagnosis.
Confirmatory Tests for VWD Diagnosis
When initial screening tests suggest a qualitative VWF defect, confirmatory tests become essential to pinpoint the specific type of VWD and guide appropriate management. These tests delve deeper into VWF function and structure.
VWF Multimer Distribution
A decreased VWF:RCo/VWF:Ag ratio should trigger evaluation of VWF multimer distribution. Multimers are different sized complexes of VWF protein, and their distribution pattern is critical for VWF function. A normal multimer distribution, in the context of a low VWF:RCo/VWF:Ag ratio, would suggest type 2M VWD. Conversely, the absence of high molecular weight multimers is a hallmark of both type 2A and type 2B VWD. Multimer assessment is typically performed using agarose gel electrophoresis [11]. This technique separates VWF multimers by size, allowing for visual analysis of their distribution. However, multimer analysis is technically demanding, time-consuming, and not routinely performed by all laboratories. Despite these limitations, it remains a valuable confirmatory test for certain VWD subtypes.
VWF Collagen Binding (VWF:CB)
Collagen binding assays (VWF:CB) play a dual role in VWF testing. Firstly, VWF:CB can serve as an indirect indicator of the presence of high molecular weight multimers. Large VWF multimers typically exhibit enhanced collagen binding. Although there is ongoing debate regarding the optimal collagen preparation for these assays [12], reduced collagen binding can suggest a deficiency in high molecular weight multimers, similar to multimer analysis. Secondly, and more specifically, some VWD variants are characterized by primary defects in VWF’s ability to bind collagen. These variants are classified as type 2M VWD. Several case reports have documented patients with borderline low VWF:Ag and VWF:RCo but significantly impaired binding to type I and/or type III collagen [13, 14]. More recently, defects in VWF binding to type VI collagen have also been reported, expanding the spectrum of collagen-related VWF defects [15]. VWF:CB assays are therefore crucial for identifying both multimer-related defects and specific collagen-binding deficiencies in VWD.
VWF–Platelet Binding (VWF:PB)
Platelet binding assays (VWF:PB) are specifically used to confirm the diagnosis of type 2B VWD. Type 2B VWD is characterized by a gain-of-function mutation in VWF that leads to increased spontaneous binding to platelets. The VWF:PB assay utilizes commercial platelets and patient plasma to directly measure this increased binding. In type 2B VWD, the assay will demonstrate elevated VWF-platelet binding compared to normal controls. Importantly, VWF:PB will not show increased binding in type 2A VWD, helping to differentiate between these two subtypes with similar initial screening test results. If VWF:PB is not available, low-dose ristocetin-induced platelet aggregation (LD-RIPA) can be used as an alternative confirmatory test for type 2B VWD.
Low-Dose Ristocetin-Induced Platelet Aggregation (LD-RIPA)
Low-dose ristocetin-induced platelet aggregation (LD-RIPA) is another valuable test for diagnosing type 2B VWD and differentiating it from platelet-type VWD. In LD-RIPA, patient platelets are exposed to low concentrations of ristocetin (typically ≤0.6 mg/mL, compared to ≥1 mg/mL in standard platelet aggregation tests). Type 2B VWD and platelet-type VWD are characterized by spontaneous platelet aggregation even at these low ristocetin concentrations. To distinguish between these two conditions, further testing such as VWF:PB or genetic analysis is required. Accurate detection of type 2B VWD is clinically important because desmopressin, a common treatment for other VWD types, is typically avoided in type 2B patients as it can exacerbate thrombocytopenia. Type 2A and type 2B VWD can otherwise present with similar patterns of decreased VWF:RCo/VWF:Ag ratio and loss of high molecular weight multimers, making LD-RIPA or VWF:PB crucial for differential diagnosis. Furthermore, differentiating platelet-type VWD is essential as its treatment strategy, unlike VWD, involves platelet transfusions.
VWF-Factor VIII Binding (VWF:FVIIIB)
The VWF-factor VIII binding assay (VWF:FVIIIB) is specifically designed to confirm the diagnosis of type 2N VWD. This assay assesses the ability of VWF isolated from patient plasma to bind to recombinant factor VIII. Type 2N VWD is defined by a specific defect in the VWF D′ and D3 domains that impairs its binding to FVIII [16]. In the VWF:FVIIIB assay, type 2N VWD patients will exhibit decreased binding of their VWF to factor VIII compared to normal controls, confirming the FVIII-binding defect.
VWF Propeptide (VWFpp)
VWF propeptide (VWFpp) measurements, specifically the VWFpp/VWF:Ag ratio, can be helpful in identifying type 1C VWD, also known as clearance defect VWD, and some type 2 VWD variants. VWFpp is a fragment cleaved from the VWF molecule during its maturation and secretion. Elevated VWFpp/VWF:Ag ratios suggest increased VWF production and turnover, which is characteristic of type 1C VWD where VWF is rapidly cleared from circulation. VWFpp is typically measured using ELISA [17]. However, it is not universally available in all laboratories. Another approach to identify clearance defects is to evaluate a patient’s response to desmopressin (DDAVP). In type 1C VWD, desmopressin administration may initially increase VWF:Ag levels at 1 hour, but these levels rapidly decline back to baseline by 4 hours due to the accelerated VWF clearance.
VWF Gene Sequencing
Genetic analysis, specifically VWF gene sequencing, has a limited but important role in VWD diagnosis. Due to the large size and complexity of the VWF gene and the high frequency of normal genetic variations (polymorphisms), routine genetic screening for VWD is not practical. Early reports linking certain sequence variations to VWD have been questioned because these ‘mutations’ are also found at a high frequency in healthy individuals [18]. However, DNA sequencing can be particularly valuable in confirming the diagnosis of type 2 VWD, especially when confirmatory laboratory tests are unavailable or when results are difficult to interpret. A substantial number of known sequence variations associated with type 2 VWD have been identified and are cataloged online by the International Society on Thrombosis and Haemostasis (ISTH).
The location of sequence variations within the VWF gene often correlates with the type of functional defect in type 2 VWD. In type 2B and type 2M VWD, where the defect involves platelet binding, mutations are commonly found in the VWF A1 domain. In type 2N VWD, characterized by impaired FVIII binding, mutations are located in the VWF D′ and D3 domains. For type 2A VWD, defects can occur in the A2 domain (near the ADAMTS13 cleavage site) or in the N- or C-terminal multimerization domains [19]. In contrast, sequence variations in type 1 and type 3 VWD are distributed throughout the VWF gene. It is crucial to interpret novel sequence variations cautiously, as not all genetic variants are necessarily pathogenic or associated with VWD.
Laboratory Classification and Diagnostic Challenges in VWD
The laboratory classification of VWD is based on the patterns of VWF abnormalities detected through the screening and confirmatory tests. Understanding these classifications is crucial for accurate diagnosis and management.
Type 1 VWD Diagnosis
Type 1 VWD, the most common form, is characterized by a mild to moderate, but proportional, decrease in both VWF:Ag and VWF:RCo. Factor VIII activity (FVIII:C) may be normal or borderline low. Bleeding symptoms in type 1 VWD are typically mild mucosal bleeding but can range to more significant surgical hemorrhage. Bleeding severity and clinical penetrance in type 1 VWD are highly variable. Several factors beyond VWF genetics influence VWF levels, including blood type, ethnicity, and modifier genes [20–22]. Physiological factors like exercise and stress can also temporarily elevate VWF levels, necessitating careful consideration and repeat testing in symptomatic patients with borderline normal VWF levels.
The diagnosis of type 1 VWD is established when both VWF:Ag and VWF:RCo are reduced, but proportionally, meaning the VWF:RCo/VWF:Ag ratio is greater than 0.6. If FVIII:C is disproportionately lower than VWF:Ag, type 2N VWD should be considered. A discrepancy between VWF:Ag and VWF:RCo, or a family history of type 2 VWD, should prompt further evaluation of VWF multimer distribution. VWFpp measurement can be helpful in investigating potential clearance defects (type 1C VWD) but is not routinely part of the diagnostic workup. Genetic testing is generally not clinically useful in type 1 VWD because not all patients have identifiable VWF gene mutations, and mutation status does not reliably predict clinical severity or treatment response. An exception is the suspicion of a clearance defect (type 1C VWD), as this diagnosis impacts treatment decisions by contraindicating desmopressin.
A major diagnostic challenge in type 1 VWD is determining the precise cutoff value for VWF:Ag and VWF:RCo below which a patient should be definitively diagnosed with VWD. Normal VWF levels exhibit wide inter-individual variation, with the lower limit of the normal range extending down to approximately 50 IU/dL (exact cutoffs vary slightly between laboratories). However, typical normal ranges inherently exclude 5% of the healthy population, meaning 2.5% of individuals would have VWF levels below the lower limit of normal by statistical definition. Since VWD prevalence is significantly less than 2.5%, low VWF levels alone are not sufficient for a VWD diagnosis. While some guidelines suggest a VWF:Ag cutoff of 30 IU/dL, others propose 20 IU/dL or 40 IU/dL [10]. The frequency of VWF gene mutations is indeed increased at lower VWF levels. Crucially, the presence and severity of bleeding symptoms must be considered in conjunction with laboratory findings. Some patients with VWF levels above a specific cutoff may still experience clinically significant bleeding requiring treatment, particularly in the context of major surgery. Therefore, a comprehensive assessment of personal and family bleeding history, with careful attention to an individual patient’s response to previous hemostatic challenges, is paramount. Laboratory findings should not be interpreted in isolation to diagnose type 1 VWD.
Type 3 VWD Diagnosis
Type 3 VWD, the most severe form, presents with distinct laboratory features including undetectable VWF:Ag and VWF:RCo, profoundly low FVIII:C, and absent VWF multimers. Patients with type 3 VWD often experience significant bleeding, including hemophilia-like joint bleeds. Some patients require prophylactic treatment to prevent recurrent bleeding, especially in the context of established joint damage. Menorrhagia is a particularly prominent and challenging symptom for women with type 3 VWD, often necessitating multiple treatment modalities for effective management.
Diagnosis of type 3 VWD is generally straightforward due to the markedly reduced or absent VWF:Ag and VWF:RCo levels. In rare instances, patients with type 1C VWD (clearance defect) may also exhibit near-undetectable VWF:Ag and VWF:RCo due to rapid VWF clearance. In such cases, VWFpp measurement can help distinguish type 1C from type 3 VWD. DNA sequencing is not typically required for diagnosis but may be useful for prenatal testing in families with known type 3 VWD.
Type 2 VWD Diagnosis
Type 2 VWD variants are characterized by a disproportionate reduction in VWF activity compared to VWF antigen levels, indicating a functional defect. These qualitative defects can affect platelet binding, factor VIII binding, or collagen binding. Bleeding symptoms in type 2 VWD are generally more severe than those seen in type 1 VWD.
Type 2A VWD Diagnosis
The combination of a decreased VWF:RCo/VWF:Ag ratio and the absence of high molecular weight multimers can be indicative of either type 2A or type 2B VWD. In type 2A VWD, VWF-platelet binding (VWF:PB) and low-dose ristocetin-induced platelet aggregation (LD-RIPA) are not increased, and patients typically do not present with thrombocytopenia. However, testing to specifically rule out type 2B VWD is crucial because desmopressin, a potential treatment option for type 2A patients, is generally contraindicated in type 2B VWD. Demonstration of a known type 2A-associated VWF gene mutation can also confirm the diagnosis, although novel genetic variants should be interpreted cautiously in the absence of clear functional data.
Type 2B VWD Diagnosis
Similar to type 2A VWD, type 2B VWD also presents with a decreased VWF:RCo/VWF:Ag ratio and a loss of high molecular weight multimers. The diagnosis of type 2B VWD is confirmed by demonstrating increased platelet binding through either an elevated VWF:PB assay or increased LD-RIPA. It’s essential to differentiate type 2B VWD from platelet-type VWD, which can have similar LD-RIPA results. Genetic sequencing can also be helpful in identifying known type 2B-associated VWF mutations. Thrombocytopenia (reduced platelet count) is frequently observed in type 2B VWD but is not a universal finding [23].
Type 2M VWD Diagnosis
Type 2M VWD encompasses patients with a decreased VWF:RCo/VWF:Ag ratio but a normal VWF multimer distribution. It can also include patients with abnormal VWF collagen binding (VWF:CB) despite having essentially normal VWF:RCo. Currently, there is no single definitive confirmatory test specifically for type 2M VWD. While gene sequencing may provide supportive evidence, novel sequence variations must be interpreted with caution, as functional consequences may be uncertain. Diagnosis often relies on excluding other type 2 variants and correlating laboratory findings with clinical bleeding history.
Type 2N VWD Diagnosis
Patients with type 2N VWD can present in two main scenarios. One involves inheriting two copies of VWF alleles with FVIII-binding defects, resulting in homozygous or compound heterozygous type 2N mutations. The severity of FVIII deficiency can vary depending on the specific mutations; some mutations lead to very low FVIII levels (around 10 IU/dL), while others result in a milder phenotype (FVIII around 20 IU/dL) [4]. These patients may have normal VWF:Ag and VWF:RCo levels but significantly reduced FVIII levels. Type 2N VWD can also occur in individuals who inherit one type 2N allele and one type 1 allele. In such cases, low VWF:Ag and VWF:RCo levels may also be observed, complicating the diagnostic picture. It is crucial to evaluate patients with isolated factor VIII deficiency (low FVIII levels with normal VWF:Ag and VWF:RCo) for type 2N VWD to avoid misdiagnosis as mild hemophilia A. Specific VWF-FVIII binding assays and genetic testing are essential for accurate diagnosis of type 2N VWD.
Conclusions
Accurate laboratory evaluation of Von Willebrand Disease is a complex process that necessitates careful integration of test results with a patient’s clinical history and bleeding symptoms. While diagnosing quantitative VWF deficiencies (type 1 and type 3 VWD) is generally more straightforward, the qualitative type 2 VWD variants present significant diagnostic challenges. Specific assays evaluating various aspects of VWF function, including platelet binding, collagen binding, and factor VIII binding, are crucial for clarifying the specific type of VWD. This precise characterization is essential to guide optimal treatment strategies and improve clinical outcomes for individuals affected by this common bleeding disorder.
Acknowledgments
The authors would like to thank the Comprehensive Center for Bleeding Disorders in Milwaukee, Wisconsin, and their fellow Zimmerman Program Investigators for thoughtful discussions on VWD, and the patients with VWD for insight into their condition.
Footnotes
CONFLICT OF INTEREST
The authors declare no conflict of interest.
References
[1] Ruggeri ZM. Structure and function of von Willebrand factor. Thromb Haemost. 1999;82(2):576-84.
[2] Wagner DD, Wagner DD. Cell biology of von Willebrand factor. Annu Rev Cell Biol. 1990;6:217-46.
[3] Lenting PJ, Vanhoorelbeke K, Vlot AJ,