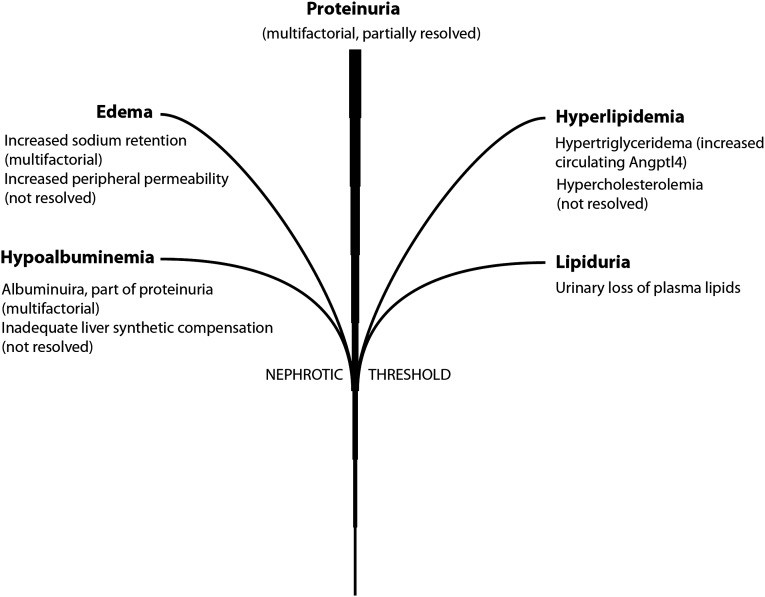
Nephrotic syndrome is a serious kidney disorder characterized by a cluster of symptoms, with proteinuria, specifically excessive protein in the urine, being the primary hallmark for diagnosis. This condition isn’t a disease itself, but rather a syndrome resulting from damage to the glomeruli, the kidney’s filtering units. Alongside significant proteinuria (typically exceeding 3.5 grams per 24 hours in adults), other key features include hypoalbuminemia (low albumin levels in the blood), edema (swelling), hyperlipidemia (high blood fats, including cholesterol and triglycerides), and lipiduria (lipids in the urine) (Figure 1). In children, nephrotic-range proteinuria is defined as urinary protein excretion rates greater than 40 mg/h per square meter of body surface area.
Nephrotic syndrome can arise from primary glomerular diseases like Minimal Change Disease (MCD), Focal Segmental Glomerulosclerosis (FSGS), and Membranous Nephropathy, or secondary to systemic conditions such as diabetes mellitus, systemic lupus erythematosus, and amyloidosis. While each component of nephrotic syndrome contributes to the overall clinical picture, proteinuria is the driving force. The other symptoms typically emerge only after protein leakage reaches a critical threshold. Understanding proteinuria and its relationship to these associated conditions is crucial for effective diagnosis and management. This article will explore the significance of proteinuria as the diagnostic hallmark and delve into the interconnected components of nephrotic syndrome.
Proteinuria: The Definitive Sign
Proteinuria, the abnormal presence of protein in the urine, is the cornerstone of nephrotic syndrome diagnosis. Healthy kidneys efficiently filter waste products from the blood while retaining essential proteins. In nephrotic syndrome, damage to the glomeruli disrupts this process, allowing significant amounts of protein, particularly albumin, to leak into the urine. This excessive protein loss is not merely a symptom; it is the initiating event that cascades into the other manifestations of the syndrome.
Several factors can contribute to glomerular damage and subsequent proteinuria. Research has identified numerous genes expressed in podocytes, specialized cells within the glomeruli, that play a critical role in maintaining the kidney’s filtration barrier. These genes are categorized based on their function, including slit diaphragm proteins, cell matrix interface proteins, cytoskeleton proteins, podocyte surface proteins, transcriptional factors, and podocyte-secreted proteins.
Figure 1: The Nephrotic Syndrome Tree. Proteinuria forms the central trunk, with branches representing edema, hyperlipidemia, hypoalbuminemia, and lipiduria, all arising from the primary issue of protein leakage.
Genetic mutations affecting proteins like nephrin and podocin, discovered through studies of congenital nephrotic syndrome and FSGS respectively, underscore the genetic underpinnings of some glomerular diseases. Transcriptional factors, such as WT1, and podocyte-secreted proteins, like Angiopoietin-like 4 (Angptl4) and vascular endothelial growth factor (VEGF), have also been implicated through genetic, animal model, and gene expression studies. Notably, a hyposialylated form of Angptl4, secreted by podocytes, has been directly linked to Minimal Change Disease, explaining many of its characteristic features.
While the precise mechanisms are still under investigation, the systemic response to nephrotic-range proteinuria is an emerging area of research. A circulating, sialylated form of Angptl4, produced by organs like adipose tissue, skeletal muscle, and the heart, has been shown to reduce proteinuria, highlighting the body’s attempt to counter protein loss. This discovery opens avenues to explore how other organs might contribute to mitigating proteinuria through secreted proteins.
Edema: Fluid Imbalance Due to Protein Loss
Edema, or swelling, is a common and visible symptom of nephrotic syndrome, developing as a consequence of hypoalbuminemia and renal salt retention. The severity and onset of edema can vary depending on the underlying cause and individual patient factors, ranging from gradual to rapid onset.
Hypoalbuminemia, a direct result of proteinuria, reduces the oncotic pressure in the blood vessels. Oncotic pressure is crucial for maintaining fluid balance by drawing fluid back into the capillaries from the tissues. When albumin levels are low, this pressure decreases, leading to fluid shifting from the bloodstream into the interstitial spaces, causing swelling, particularly in the legs, ankles, and around the eyes.
Furthermore, the kidneys in nephrotic syndrome often retain sodium and water, exacerbating fluid overload and edema. This renal salt retention is targeted by diuretic medications, which work on different parts of the nephron (thick ascending loop of Henle, distal tubule, collecting duct, and proximal tubule) to promote sodium and water excretion. While albumin infusions might be used in severe cases of edema, diuretics are the mainstay of treatment. Increased peripheral capillary permeability is another contributing factor to edema in nephrotic syndrome, though it is less understood and not directly targeted by current therapies.
Hypercholesterolemia: Disrupted Lipid Metabolism
Hypercholesterolemia, elevated cholesterol levels, is another hallmark component of nephrotic syndrome. Patients typically exhibit high total and LDL cholesterol, primarily due to a deficiency in LDL receptors, which are responsible for removing cholesterol from the circulation. This impaired cholesterol removal triggers the liver to compensate by increasing cholesterol production.
The liver’s response involves upregulation of enzymes like 3-hydroxy-3-methyl glutaryl-CoA reductase (HMG-CoA reductase), a key enzyme in cholesterol biosynthesis, and Acyl-CoA cholesterol acyltransferase-2 (ACAT2), involved in cholesterol esterification. Despite normal LDL receptor mRNA levels in the liver, suggesting a post-transcriptional issue, the exact mechanisms are still being investigated. Recent research points to increased hepatic degradation of LDL receptors by proteins like proprotein convertase subtilisin/kexin type 9 (PCSK9) and inducible degrader of the LDL receptor (IDOL) as potential contributors. Additionally, urinary loss of proteins like lecithin-cholesterol acyltransferase (LCAT) may further contribute to hypercholesterolemia.
Hypertriglyceridemia: Impaired Triglyceride Clearance
Hypertriglyceridemia, elevated triglyceride levels, in nephrotic syndrome arises from impaired clearance of triglycerides from the blood. This is largely attributed to the inactivation of lipoprotein lipase (LPL), an enzyme that breaks down triglycerides, by Angptl4.
Circulating Angptl4, while playing a role in reducing proteinuria, also has the side effect of inhibiting LPL activity. Angptl4 converts the active dimeric form of LPL into inactive monomers, reducing the conversion of triglycerides into free fatty acids. Both LPL dimers and monomers are lost in the urine in nephrotic syndrome, further contributing to the dysregulation of lipid metabolism.
Interestingly, proteinuria and hypertriglyceridemia are interconnected through negative feedback loops involving Angptl4 and free fatty acids, as elaborated below.
Hypoalbuminemia: Loss and Insufficient Production of Albumin
Hypoalbuminemia, low levels of albumin in the blood, is a direct consequence of urinary albumin loss in nephrotic syndrome. While the liver can normally increase albumin production, it often fails to fully compensate for the significant protein losses in nephrotic syndrome.
Albumin plays a crucial role in maintaining oncotic pressure and also acts as a carrier for free fatty acids (FFAs) in the blood. Albumin has multiple binding sites for FFAs. In nephrotic syndrome, there’s a preferential loss of albumin with lower FFA content in the urine, leading to a relative accumulation of albumin with higher FFA content in the circulation. This, coupled with reduced overall albumin levels, results in an elevated plasma FFA-to-albumin ratio.
This increased FFA-to-albumin ratio drives FFA uptake by skeletal muscle, heart, and adipose tissue, contributing to hypertriglyceridemia and triggering a systemic response to reduce proteinuria through Angptl4 secretion.
Lipiduria: Lipids in the Urine
Lipiduria, the presence of lipids in the urine, is generally considered secondary to hyperlipidemia. It mainly results from the filtration of high-density lipoprotein (HDL) particles, which are smaller in size and can pass through the damaged glomerular filter.
These lipids can be observed in the urine as oval fat bodies (tubular cells containing lipids), fatty casts, or free-floating lipid globules. Lipid components rich in esterified cholesterol exhibit a characteristic Maltese cross appearance under polarized light microscopy, which can be a diagnostic clue.
Molecular Interplay: Proteinuria and Hypertriglyceridemia
The molecular link between proteinuria and hypertriglyceridemia is one of the most well-understood connections in nephrotic syndrome. It involves a complex interplay between free fatty acids (FFAs), albumin, and Angptl4, creating both local and systemic feedback loops (Figure 2 and Figure 3).
Under normal conditions, skeletal muscle, heart, and adipose tissue utilize FFAs as a primary energy source, derived from both LPL-mediated hydrolysis of circulating triglycerides and albumin-bound FFAs. These organs express LPL, Angptl4, and PPARs (peroxisome proliferator-activated receptors), which regulate Angptl4 expression in response to FFA uptake.
In nephrotic syndrome, the preferential urinary loss of albumin with low FFA content leads to a higher proportion of albumin with high FFA content remaining in circulation. This shifts the FFA uptake balance towards albumin-bound FFAs. Increased uptake of albumin-bound FFAs in peripheral tissues triggers local Angptl4 upregulation, likely mediated by PPARs. This locally produced Angptl4 then inactivates LPL in the same tissues, reducing triglyceride breakdown and contributing to hypertriglyceridemia.
Figure 2: FFA Sources in Normal and Nephrotic States. This diagram contrasts the balance of FFA sources in healthy individuals (green) with the altered balance in nephrotic syndrome (red), where albumin-bound FFAs become dominant.
However, this local feedback loop is intertwined with a larger systemic feedback loop aimed at reducing proteinuria. Angptl4 released from skeletal muscle, heart, and adipose tissue enters the circulation and binds to glomerular endothelial αvβ5 integrin, reducing proteinuria. Elevated plasma Angptl4 levels are observed in patients with various causes of nephrotic syndrome, indicating this systemic response.
Figure 3: Negative Feedback Loops in Nephrotic Syndrome. This schematic details the interconnected feedback loops involving proteinuria, hypoalbuminemia, hypertriglyceridemia, Angptl4, and FFA, illustrating both the local and systemic effects of Angptl4.
Nephrotic-Range Proteinuria Threshold: A Molecular Perspective
The concept of a nephrotic-range proteinuria threshold, typically around 3.5 g/day in adults, highlights a critical point beyond which other nephrotic syndrome components become clinically evident. The molecular basis for this threshold, in the context of hypertriglyceridemia, is linked to the plasma FFA-to-albumin ratio.
Studies in animal models show that during mild proteinuria, the plasma FFA-to-albumin ratio, plasma Angptl4 levels, and peripheral organ Angptl4 and PPAR mRNA expression remain similar to healthy controls. However, in severe, nephrotic-range proteinuria, these parameters significantly increase. This suggests that the nephrotic threshold, at least concerning hypertriglyceridemia, is associated with the downstream effects of an elevated plasma FFA-to-albumin ratio, triggering the Angptl4-mediated feedback loops.
Conclusion: Proteinuria as the Defining Diagnostic Hallmark
In conclusion, while nephrotic syndrome is characterized by a constellation of symptoms including edema, hyperlipidemia, hypoalbuminemia, and lipiduria, proteinuria remains the definitive diagnostic hallmark. The excessive loss of protein in the urine is not only a key indicator but also the primary driver of the syndrome’s other manifestations.
Understanding the molecular mechanisms underlying proteinuria and its intricate relationships with other components is crucial for developing targeted therapies. The study of Angptl4, in particular, has revealed potential therapeutic strategies for managing proteinuria and associated lipid abnormalities. Future research focusing on the circulating glomerulophilic proteome may unlock further novel treatments for proteinuric kidney diseases, offering hope for improved management and outcomes for individuals with nephrotic syndrome.
References
[1] … (References from the original article would be listed here, maintaining the numbering and content)