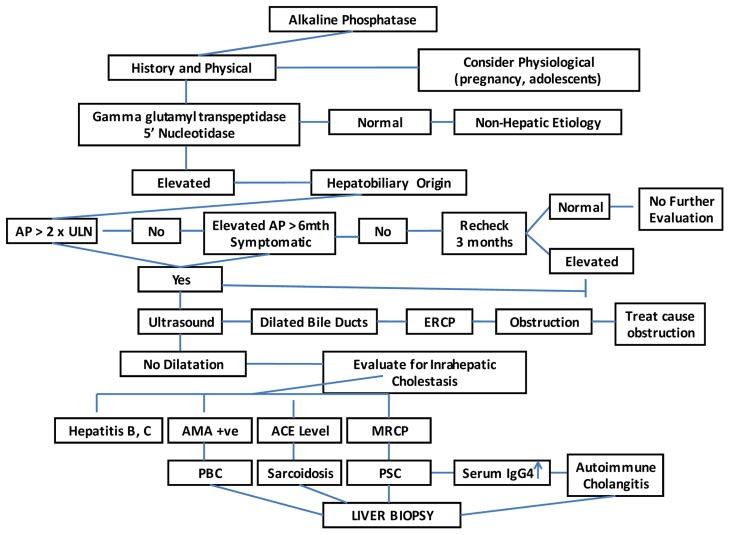
Elevated serum alkaline phosphatase (AP) is a common clinical finding that often presents a diagnostic challenge. Cholestasis, a condition arising from impaired bile formation, secretion, or flow obstruction, frequently manifests with disproportionately elevated AP and gamma-glutamyltransferase (GGT) relative to aminotransferases. This article delves into the differential diagnosis of raised alkaline phosphatase, particularly in the context of cholestasis, providing a detailed overview for accurate diagnosis and management.
Understanding Cholestasis and Alkaline Phosphatase
Cholestasis, derived from the Greek words “chole” (bile) and “stasis” (standing still), indicates a stagnation of bile. This can stem from issues in bile synthesis within liver cells, secretion processes, or blockages in the bile ducts, either inside or outside the liver. Chronic cholestatic disorders often present with pruritus (itching) and fatigue as primary symptoms. Diagnosing these conditions necessitates visualizing the biliary tree through cholangiography and examining liver tissue via histology. Advances in understanding the genetic and immunological underpinnings of cholestasis are paving the way for innovative treatments.
Representative Case
Consider a 68-year-old man experiencing intermittent right upper quadrant pain for two years, described as a dull ache. He also noted a 20-pound weight loss, dark urine, and pale stools. He denied medication use or excessive alcohol intake and had no significant medical history. Physical examination revealed jaundice and mild right upper quadrant tenderness. Lab results showed elevated total bilirubin (2.7mg/dl), AP (437 U/L), ALT (85 U/L), and AST (54 U/L). A CT scan of the abdomen indicated mild extrahepatic bile duct prominence and patchy intrahepatic ductal dilation. ERCP confirmed multifocal intrahepatic biliary strictures with beading and dilation, suggestive of sclerosing cholangitis. Cytology was negative for malignancy. Liver biopsy showed moderate to marked portal inflammation with lymphocytes and plasma cells. IgG4 immunostaining revealed up to 20 IgG4-positive plasma cells per high power field, and serum IgG4 was 320mg/dl. The diagnosis was IgG4-related autoimmune cholangitis, and corticosteroid therapy was initiated.
The Physiology of Bile Formation and Alkaline Phosphatase Elevation
Bile formation is driven by bile acids, the primary organic components of bile. Hepatocytes, liver cells, transport conjugated bile acids across their basolateral membrane using two key transporters: Na+-dependent NTCP (Sodium Taurocholate Co-Transporting polypeptide, SLC10A1) and Na+-independent OATPs (organic anion transporting proteins OATP2, SLC21A6). NTCP, found exclusively in hepatocytes, handles both conjugated and unconjugated bile acids. OATPs transport various organic anions, bile salts, and xenobiotics. These transporters are crucial for bile synthesis and the liver’s detoxification processes, eliminating bile salts, bilirubin, cholesterol, and drugs.
Bile acid efflux into bile is facilitated by ATP-binding cassette (ABC) transporters on the hepatocyte canalicular membrane. The primary transporter is BSEP (bile salt export pump, ABCB11). Other ABC transporters include MRP2 (ABCC2, for bilirubin and other organic anions), MDR1 (ABCB1, for organic cations), and MDR2 (ABCB4, phospholipid export). MRP2 also detoxifies drugs and toxins. MDR3 (ABCB4) is unique in flipping phosphatidylcholine, a key lipid, into bile. The expression of these transporters is tightly regulated to prevent bile acid toxicity in hepatocytes.
Once secreted, bile undergoes changes as it moves through the biliary tree. Cholangiocytes, bile duct cells, reabsorb bile salts via ASBT (SLC10A2), enabling intrahepatic bile salt cycling. Cholangiocyte transporters like AE2 (SLC10A2) and CFTR (ABCC7) modify bile composition. Intestinal reabsorption of bile salts via ASBT in the ileum, followed by efflux into portal circulation via MRP3, is critical for bile acid homeostasis and enterohepatic circulation. Nuclear receptors (NRs) like FXR, PXR, VDR, and CAR regulate bile acid metabolism. During cholestasis, FXR and PXR activation reduces bile acid synthesis by downregulating CYP7A1. FXR upregulates BSEP and downregulates NTCP, while PXR activates Oatp2 and CAR regulates MRP2 and MPR3. This coordinated NR activation reduces bile acid synthesis and uptake while increasing efflux, protecting hepatocytes from toxic bile salt accumulation.
Alkaline phosphatase (AP) is an enzyme present in various tissues, including liver, bone, intestine, kidney, placenta, and leukocytes. Elevated serum AP is often associated with liver or bone disorders. In cholestasis, AP elevation is thought to arise from increased synthesis and release from bile duct epithelial cells due to bile flow obstruction. While AP is a marker of cholestasis, it’s not liver-specific, necessitating further investigation to pinpoint the cause of elevation.
Liver Test Patterns in Cholestasis and Elevated Alkaline Phosphatase
Liver function tests (LFTs), although misnamed as they don’t directly measure liver function, are crucial in assessing liver health. They include serum liver enzymes (aminotransferases, AP) and function markers (prothrombin time, bilirubin, albumin). Abnormal LFT patterns can indicate hepatocellular, cholestatic, mixed, or infiltrative liver disease, aiding in differential diagnosis.
| Test | Hepatocellular | Cholestatic | Infiltrative | Mixed |
|---|---|---|---|---|
| ALT/AST | ++ | N/+ | N/+ | ++ |
| AP | N/+ | ++ | +/++ | ++ |
| Total Bilirubin | N/+ to ++ | N/+ to ++ | N/+ | N/+ to ++ |
ALT – alanine aminotransferase; AST – aspartate aminotransferase; AP – alkaline phosphatase; TB – total bilirubin, N – normal; + to ++ – degree of elevation
Persistent elevation of serum AP presents a diagnostic challenge. While high concentrations are in liver and bone, AP also exists in other organs. Serum AP levels vary with age, gender, and blood type. Mild elevations are normal in infants, puberty, and ages 40-65, particularly in women. Adolescent boys may have 2-5 times higher AP levels due to bone growth. African Americans and smokers may also have slightly higher AP. Blood types O and B can show elevated AP after fatty meals due to intestinal AP influx. Understanding these physiological variations is vital when interpreting raised alkaline phosphatase levels.
Non-Hepatic Causes of Elevated Alkaline Phosphatase
It’s crucial to consider non-hepatic causes when evaluating elevated AP.
| Physiological | Pregnancy, Adolescence, Post-fatty meal (blood groups O or B) |
|---|---|
| Bone Disease | Healing Fracture, Paget’s Disease, Osteomalacia, Vitamin D insufficiency, Rickets, Malignancy (osteogenic sarcoma, metastatic) |
| Renal | Renal Failure |
| Heart | Heart Failure |
| Endocrine | Hyperthyroidism, Hyperparathyroidism |
| Malignancy | Lymphoma, Leukemia, Renal cell carcinoma, Multiple endocrine neoplasia (MEN) II |
These conditions can raise AP levels independently of liver disease, necessitating careful clinical correlation.
Cholestatic Liver Diseases: Intrahepatic and Extrahepatic Causes
Cholestatic liver diseases are broadly categorized into intrahepatic and extrahepatic causes. Understanding this distinction is crucial for differential diagnosis of raised alkaline phosphatase.
| INTRAHEPATIC CHOLESTASIS | EXTRAHEPATIC CHOLESTASIS |
|---|---|
| Hepatitis: Viral (B,C), Alcoholic | Extrinsic Obstruction |
| Genetic: BRIC, PFIC, Dubin-Johnson, Rotor’s | – Stones |
| Drugs and Herbal Remedies | – Malignancy (Pancreas, Gallbladder, Metastatic, Cholangiocarcinoma, Ampullary cancer) |
| Pregnancy (ICP) | Pancreatitis, Pancreatic pseudocyst, Parasitic Infection |
| PBC, PSC | Secondary sclerosis (surgery, chemotherapy) |
| Granulomatous liver disease (Infections, Sarcoidosis) | |
| Infiltrative: Amyloidosis, Lymphoma | |
| Idiopathic Adult Ductopenia | |
| Autoimmune Cholangitis (PSC-like), Cholangiopathy (PBC-like) | |
| Prolonged TPN, Postoperative state, Sepsis, Malignancy (Hepatocellular, Metastatic) |
Intrahepatic cholestasis arises from within the liver, affecting bile ducts or hepatocytes. Extrahepatic cholestasis results from obstruction outside the liver, typically in the larger bile ducts.
Causes of Low Alkaline Phosphatase
While elevated AP is more common, low AP levels can also occur, though less frequently, and are associated with specific conditions.
| Malnutrition |
|---|
| Wilson’s Disease |
| Hypothyroidism |
| Zinc Deficiency |
| Vitamin C Deficiency |
| Low Phosphorus Level |
| Pernicious Anemia |
Low AP is less commonly investigated in the context of cholestasis but should be considered in relevant clinical settings.
Evaluating Cholestasis and Differential Diagnosis
The initial step in evaluating elevated AP is to identify its source. Fractionation of AP isoenzymes via electrophoresis or measuring 5’nucleotidase and GGT levels can help, as the latter two are elevated in hepatobiliary disease. Next, it’s crucial to determine if cholestasis is intrahepatic or extrahepatic. Intrahepatic cholestasis involves bile excretion impairment at the hepatocellular level, while extrahepatic cholestasis involves large bile duct issues due to intrinsic processes or external compression. Conditions like primary sclerosing cholangitis (PSC) can affect both intrahepatic and extrahepatic ducts.
History and Physical Examination
A detailed medical history and physical examination are paramount. Painless jaundice with a palpable mass suggests malignant biliary obstruction, while choledocholithiasis often presents with abdominal pain and jaundice. Acute ascending cholangitis, a life-threatening complication of common bile duct stones, requires urgent intervention and may present with fever, right upper quadrant pain, and jaundice (Charcot’s triad). Prior biliary surgery or manipulations increase cholangitis risk. History should include alcohol and drug use, medications (including herbal and over-the-counter), and relevant medical conditions. The clinical setting, such as cholestasis in a critically ill patient (likely sepsis-related), is important. Family history of cholestatic liver disease may indicate a hereditary disorder.
Pruritus and fatigue are common, though non-specific, symptoms of intrahepatic cholestasis, also seen in extrahepatic cholestasis and other liver diseases. Severe pruritus can significantly impair quality of life and may necessitate liver transplantation. In infants, pruritus manifests as irritability and failure to thrive, while older children may experience school and sleep issues. The exact mechanism of pruritus in liver disease is unclear, possibly related to bile salt retention or endogenous opioids. Fatigue is another significant, often underrecognized symptom in chronic cholestatic liver diseases like primary biliary cirrhosis (PBC).
Hypercholesterolemia is frequently observed in intrahepatic cholestasis, often with lipoprotein X, which has low atherogenic potential. Lipid abnormalities can manifest as xanthomas (cholesterol deposits in tendons, bony prominences, nerves) or xanthelasmas (deposits in periorbital skin folds). Fat-soluble vitamin (A, D, E, K) malabsorption is common, requiring careful management to prevent deficiencies. Osteopenia and osteoporosis are also frequent complications of chronic cholestatic liver disease.
Investigations for Differential Diagnosis
Initial laboratory investigations should include a complete blood count, liver enzymes (aminotransferases, AP, GGT), liver synthetic function tests (albumin, bilirubin, prothrombin time), viral hepatitis serologies, autoantibodies (ANA, SMA, AMA, p-ANCA), and immunoglobulin levels.
Transcutaneous abdominal ultrasound (TUS) is typically the first imaging modality to assess for biliary dilatation. It’s non-invasive, inexpensive, and specific for bile duct obstruction. However, it can be technically challenging in obese patients, and may miss common bile duct (CBD) stones. CT scans are comparable to TUS for stone detection and offer additional information about liver parenchyma and mass lesions, but are less effective for biliary tree delineation. If extrahepatic obstruction is suspected despite negative TUS and CT, further imaging is needed. Magnetic resonance cholangiopancreatography (MRCP) and endoscopic ultrasound (EUS) are accurate for detecting extrahepatic obstruction. Endoscopic retrograde cholangiopancreatography (ERCP) is the gold standard for visualizing the biliary tract, identifying obstruction causes and levels, and enabling interventions like stone extraction, stricture dilation, and stenting. Percutaneous transhepatic cholangiography may be necessary in patients with altered anatomy.
For suspected intrahepatic cholestasis, specific autoantibodies are informative. A positive antimitochondrial antibody (AMA) is highly suggestive of PBC. Diagnostic criteria for PBC include elevated AP, positive AMA, and characteristic liver biopsy findings, although biopsy is not always essential. Liver histology resembling PBC with biochemical features of PBC but autoantibodies suggesting autoimmune hepatitis (positive ANA or ASMA) is termed autoimmune cholangiopathy. Low titers of ANA and SMA can be seen in PBC, PSC, and other cholestatic conditions. In AMA-negative cases, MRCP is indicated to rule out PSC, characterized by multifocal strictures and dilatations of bile ducts. An abnormal cholangiogram resembling PSC in a relevant clinical context may suggest AIDS cholangiopathy or secondary cholangitis. Liver biopsy is indicated when the diagnosis is unclear or for staging, requiring at least 10 portal tracts for accurate diagnosis.
Genetic Disorders and Raised Alkaline Phosphatase
Genetic disorders affecting hepatocellular transport systems can lead to cholestasis. These include progressive familial intrahepatic cholestasis (PFIC), benign recurrent intrahepatic cholestasis (BRIC), cystic fibrosis, and Dubin–Johnson syndrome.
| Protein/Gene | Chromosome | Clinical Disease State | Lab Findings | Characteristic Features |
|---|---|---|---|---|
| FIC1/ATP8B1 | 18q21 | PFIC1 | GGT low, Bile acids increased | AR; severe pruritus; cholestasis; no ductular proliferation |
| BRIC1 | GGT low | Periodic cholestasis attacks | ||
| BSEP/ABCB11 | 2q24 | PFIC2 | GGT low, Bile acids increased | AR; severe pruritus; cholestasis; no ductular proliferation |
| BRIC2 | GGT low | Periodic cholestasis attacks | ||
| ICP | GGT low or high | Cholestasis, pruritus in 3rd trimester pregnancy | ||
| MDR3/ABCB4 | 7q21 | PFIC3 | GGT increased, Bile acids normal | AR; moderate pruritus; cholestasis |
| ICP | ||||
| MRP2/ABCC2 | 10q24 | Dubin Johnson | Conjugated hyperbilirubinemia | Jaundice |
| CFTR/ABCC7 | 7q31 | Cystic Fibrosis | Associated PSC |
PFIC encompasses autosomal recessive cholestatic liver diseases often presenting in infancy. PFIC1 (Byler Disease) and PFIC2 are similar, caused by mutations in FIC1 and BSEP genes, respectively, leading to severe cholestasis, pruritus, elevated bile acids, and normal GGT. PFIC2 has higher aminotransferases and hepatocellular carcinoma risk. PFIC1 may involve diarrhea, bile acid malabsorption, pancreatitis, and nephrolithiasis. PFIC3, due to ABCB4 mutations, shows elevated GGT and presents later in infancy or childhood, potentially leading to liver failure. MDR3 heterozygous women have increased ICP risk.
BRIC is a rare condition with intermittent cholestasis episodes. BRIC1 and BRIC2 are due to FIC1 and BSEP mutations, respectively, with benign courses. Cholestatic episodes show elevated bilirubin and AP, but normal GGT. Dubin-Johnson syndrome, caused by MRP2 mutations, presents with conjugated hyperbilirubinemia and elevated GGT. Cystic fibrosis, due to CFTR mutations, can involve cholestasis. Alagille’s syndrome, from JAG1 mutations, features bile duct hypoplasia and developmental defects.
Primary Biliary Cirrhosis (PBC)
PBC, or chronic nonsuppurative destructive cholangitis, has unknown etiology but is believed to involve genetic susceptibility and environmental triggers. Genetic factors are suggested by high concordance in monozygotic twins and familial occurrence. HLA-DR8 and HLA-DPB1 links suggest immune system abnormalities. Xenobiotics and infections may trigger autoimmune reactions or direct toxicity. Smoking is a possible risk factor and may accelerate progression.
Clinical Features and Diagnosis of PBC
PBC is more common in women (10:1 female:male ratio). Approximately 60% are asymptomatic at diagnosis, detected incidentally by elevated AP. Fatigue and pruritus are common symptoms but don’t correlate with disease severity. Fat-soluble vitamin deficiencies and osteoporosis occur in advanced PBC.
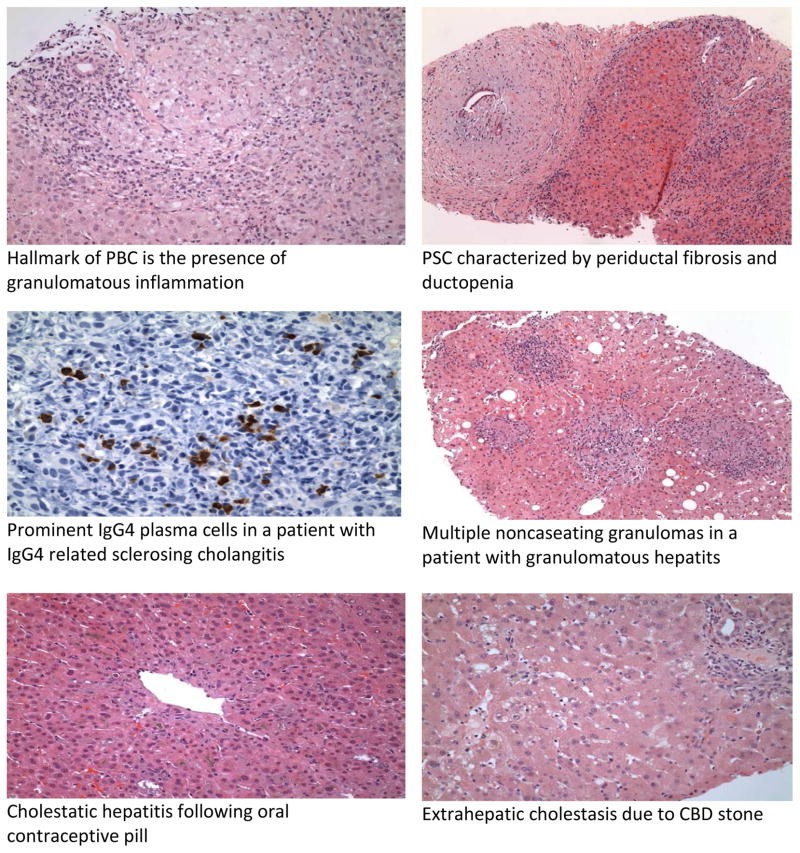
Diagnosis is characterized by cholestatic liver enzyme abnormalities with elevated AP and GGT. Serum bilirubin is a prognostic indicator. The hallmark of PBC is a positive AMA, present in 90-95% of patients. ANA may be present in 20-50%. Imaging excludes biliary obstruction and screens for cirrhosis and hepatocellular carcinoma in advanced cases. Liver histology shows “florid duct lesions” with bile duct inflammation and injury, comprising plasma cells, macrophages, and eosinophils, and potentially noncaseating granulomas. Ductopenia may be the sole histological finding.
Treatment of PBC
Ursodeoxycholic acid (UDCA) is the primary FDA-approved treatment for PBC, improving liver tests and histology, and slowing disease progression, especially in early stages. It reduces liver transplant needs and mortality in responsive patients. Liver transplantation is for decompensated PBC. Post-transplant PBC recurrence occurs in about 20% despite persistent AMA positivity.
Autoimmune Cholangiopathy (AIC)
AIC describes AMA-negative PBC (5-8% of PBC cases), characterized by cholestatic liver enzymes, PBC-consistent histology, and negative AMA. Hypergammaglobulinemia and positive ANA/SMA may be present. AIC is treated like PBC and might represent a PBC variant with autoantibody profile variations.
| PBC | AIC | PSC | IgG4-SC | |
|---|---|---|---|---|
| ANA | 20–50% | 79–100% | 7–77% | 43–80% |
| SMA | 20% | 50% | 15–20% | |
| AMA | 95% | 0 | 0 | 0 |
| p-ANCA | 3–33% | Unknown | 80% | |
| Ig | IgM | IgG | IgG, IgM | IgG |
PBC –primary biliary cirrhosis; AIC- autoimmune cholangiopathy; PSC – primary sclerosing cholangitis; IgG4-SC – IgG4 relates sclerosing cholangitis; ANA – anti nuclear antibody; SMA – smooth muscle antibody; p-ANCA perinuclear anti-neutrophilic cytoplasmic antibody; Ig – immunoglobulin
Primary Sclerosing Cholangitis (PSC)
PSC is a chronic cholestatic liver disease featuring fibrosis of intrahepatic and/or extrahepatic bile ducts, potentially progressing to biliary cirrhosis. It is strongly linked to inflammatory bowel disease (IBD), particularly ulcerative colitis (UC), affecting 62-73% of PSC patients. Despite IBD being more common in women, PSC is more prevalent in men (2:1 male:female). Median onset age is around 40 years, and it’s more common in non-smokers.
PSC’s etiology is multifactorial, involving genetic predisposition and environmental factors. Siblings of PSC patients have a significantly higher risk. HLA-B8, HLA-DR3, and HLA-DRw52a associations are noted. Genome-wide association studies (GWAS) have identified non-HLA susceptibility loci also implicated in UC. The PSC-UC association suggests autoimmune and infectious contributions. Autoantibodies like p-ANCA or ANA are present, but unlike other autoimmune conditions, PSC is male-predominant and corticosteroid-resistant.
Clinical Features and Diagnosis of PSC
PSC should be suspected in IBD patients with cholestatic liver enzyme abnormalities. Most patients are asymptomatic initially. Common symptoms include intermittent fatigue, pruritus, and abdominal discomfort, often resolving spontaneously. Serum AP and bilirubin may fluctuate. Fever, chills, and jaundice suggest bacterial cholangitis due to biliary obstruction. Persistent jaundice raises suspicion of cholangiocarcinoma (CCA).
ERCP was the gold standard for PSC diagnosis, but MRCP is increasingly used as a non-invasive alternative. ERCP is reserved for interventions like stricture dilation or when MRCP is suboptimal, or to obtain cytology for CCA from dominant strictures. Cholangiography shows short strictures alternating with dilated segments, creating a “beaded” appearance. Histology may show “onion skin” fibrosis (periductal concentric fibrosis) in 7-50% of cases, ductopenia, and bile duct proliferation. Obliterative fibrous cholangitis is less common. Secondary sclerosing cholangitis causes must be excluded.
PSC patients have a 5-10% lifetime CCA risk. Other malignancy risks include gallbladder, colorectal, and hepatocellular cancers. Annual MRI/MRCP or ultrasound with CA19-9 is recommended for CCA screening, and annual colonoscopy with biopsies for colorectal cancer screening.
Treatment of PSC
UDCA has been studied for PSC treatment with inconclusive results at low doses and adverse effects at high doses. UDCA (13-15 mg/kg/day) may improve cholestasis features but lacks survival benefit or transplant delay, and is not generally recommended for PSC treatment.
IgG4-Related Sclerosing Cholangitis (IgG4-SC)
IgG4-related sclerosing disease is a systemic inflammatory condition with IgG4-positive plasma cells and T-lymphocyte infiltration, often affecting multiple organs, including pancreas, bile ducts, gallbladder, kidney, and salivary glands. IgG4-SC is a variant involving bile ducts and is associated with autoimmune pancreatitis (AIP) in 80-90% of cases.
Clinical Features and Diagnosis of IgG4-SC
IgG4-SC clinically and cholangiographically resembles PSC but is steroid-responsive.
| Feature | PSC | IgG4-SC |
|---|---|---|
| Age | 25–45 yrs | 65 yrs |
| Gender | 65% Male | 80% Male |
| IBD Association | Present | Absent |
| Other Organ Involvement | No | Pancreas frequently involved |
| p-ANCA | Positive | Less common |
| Elevated Serum IgG4 | 7–9% | 70% |
| Histology | Ductopenia, Periductal concentric fibrosis | Abundant IgG4+ plasma cells, Periportal fibroinflammatory nodules |
| Cholangiogram | Multifocal “beaded”; pruned tree appearance of intrahepatic and extrahepatic ducts | Segmental, long strictures with prestenotic dilatation, distal CBD involvement, pancreatic duct involvement |
| Response to Steroids | No | Yes |
IgG4-SC diagnosis is easier with associated AIP, which presents with “sausage-like” pancreatic swelling, pancreatic duct narrowing, and elevated serum IgG4. IgG4 levels >140mg/dl in AIP show high sensitivity and specificity. Cholangiographic features are classified into 4 types based on stricture location. Histology is the gold standard, showing lymphoplasmacytic infiltration, IgG4+ plasma cells, and obliterative phlebitis, distinct from PSC’s ductopenia and periductal fibrosis.
Treatment of IgG4-SC
IgG4-SC is steroid-responsive, inducing remission. Initial prednisone 40mg/day for 3-4 weeks, tapered based on response, is typical. Steroid-sparing agents like azathioprine, mycophenolate mofetil, and rituximab may be used for relapsing cases requiring chronic immunosuppression.
Hepatic Sarcoidosis
Sarcoidosis is a multisystemic disease of unknown cause, characterized by non-caseating granulomas, primarily affecting lungs and lymph nodes, but also the liver. Hepatic sarcoidosis typically affects individuals aged 20-40 years, with higher prevalence and severity in African Americans.
Clinical Features and Diagnosis of Hepatic Sarcoidosis
Most hepatic sarcoidosis patients are asymptomatic (50-80% with systemic sarcoidosis have liver involvement), while 5-30% present with nausea, vomiting, abdominal pain, fever, night sweats, myalgia, and weight loss. Elevated AP and GGT are common biochemical abnormalities, reflecting fibrosis severity. Serum angiotensin-converting enzyme (ACE) is often elevated in active hepatic sarcoidosis. Hepatic nodules are usually small, multiple, and diffuse. CT and MRI show low-attenuating, non-enhancing lesions. Histology reveals noncaseating granulomas, typically in portal and periportal areas, with multinucleated giant cells. Advanced cases may show chronic cholestasis, ductopenia, and cirrhosis. Granulomas are not specific to sarcoidosis and can be seen in other conditions.
Treatment of Hepatic Sarcoidosis
Corticosteroids are the mainstay of treatment. Treatment is generally not needed without symptoms or biochemical abnormalities. Low-dose prednisone (10-15mg/day) can improve symptoms and liver tests in symptomatic patients with mild to moderate enzyme elevation, but biochemical response doesn’t always correlate with histologic improvement, and cirrhosis can progress. Advanced hepatic sarcoidosis may not respond to steroids. Steroid-sparing agents like methotrexate, hydroxychloroquine, azathioprine, and cyclophosphamide have shown benefit. UDCA may be useful in chronic cholestasis. Portal hypertension, a rare complication, can occur with or without cirrhosis and may require liver transplantation.
Parenteral Nutrition-Associated Liver Disease (PNALD)
PNALD is more common in children, especially premature infants, on parenteral nutrition (PN). It should be considered in PN patients with hyperbilirubinemia and liver enzyme elevation after excluding other causes. Liver enzyme elevation usually appears within 1-4 weeks of PN initiation and often improves despite continued PN. Injury patterns include steatosis/steatohepatitis and cholestasis. Steatosis is more common in adults, cholestasis in children. PNALD severity ranges from mild enzyme elevation to severe hepatobiliary damage and cirrhosis.
Etiology is multifactorial, involving decreased gallbladder emptying, bacterial overgrowth, and PN amino acid composition. Biliary system immaturity is a major factor in infant cholestasis. PNALD prevention includes early enteral feeding, hypoxia prevention, sepsis treatment, hypoproteinemia management, and avoiding hepatotoxic drugs. Cyclic PN and reduced amino acid content may also help.
Treatment of PNALD
PNALD treatment options are limited. UDCA may be effective. Bowel decontamination with antibiotics like metronidazole may be helpful. Intestinal transplantation, often with liver transplantation, is reserved for PN-dependent short bowel syndrome patients with overt or pending liver failure.
Intrahepatic Cholestasis of Pregnancy (ICP)
ICP is defined by pruritus and elevated serum bile acid levels in pregnant women. It usually starts in the late second or third trimester, resolves after delivery, and recurs in subsequent pregnancies.
Clinical Features and Diagnosis of ICP
Pruritus, often worse at night, is the main symptom. Steatorrhea can occur. Jaundice is less common. Hormonal, genetic (MDR3 mutations in ~15% of cases), and environmental factors contribute. Diagnosis is by exclusion after ruling out other hepatic impairments. Serum AP is elevated but less diagnostically useful in pregnancy. Aminotransferase elevation is mild to moderate, and bilirubin is usually <5 mg/dL. Serum bile acid level, particularly cholic acid, is a sensitive and specific marker for diagnosis and monitoring.
Treatment of ICP
UDCA is the treatment of choice for ICP, controlling symptoms and reducing bilirubin and bile acid levels without maternal or fetal adverse effects. Dexamethasone is less effective than UDCA.
Cholestasis Related to Sepsis
Sepsis is a common cause of jaundice and cholestasis in the ICU, often linked to bacterial infections like E. coli. Sepsis-induced liver disease can be primary (hypotension/hypoxia-related, with high aminotransferases) or secondary (Kupffer cell activation, inflammatory mediators, with cholestasis).
Intrahepatic cholestasis in sepsis shows disproportionate bilirubin elevation compared to AP. AP is typically 2-3 times normal, and aminotransferases are <2 times normal. Biliary obstruction and infection must be excluded. Pruritus is not prominent. Histology shows bile in canaliculi and hepatocytes, Kupffer cell hyperplasia, and apoptotic bodies. Treatment involves aggressive supportive care and infection management. UDCA use is not well-established in sepsis-related cholestasis.
Drug-Induced Liver Disease (DILI)
DILI is a common cause of cholestatic liver disease, accounting for 40% of adult hepatitis cases, and is triggered by prescription, over-the-counter, herbal medications, and toxins. DILI can manifest as pure cholestasis or mixed hepatocellular-cholestatic patterns. Pathogenesis involves direct drug toxicity, immune responses, and hepatobiliary transporter inhibition. ABCB11 and ABCB4 gene mutations may increase DILI susceptibility.
Clinical Features and Diagnosis of DILI
DILI symptoms are nonspecific (abdominal pain, nausea, fatigue) or acute (jaundice). Chronic DILI can present with pruritus. Resolution often follows drug withdrawal, but chronic liver disease can occur. DILI should be suspected in unexplained liver test abnormalities after medication initiation, with improvement upon drug cessation.
| Clinical Characteristics | Examples |
|---|---|
| Subclinical | Sulfonamide, Salicylate |
| Acute Liver Injury | Acetaminophen (hepatocellular), Amoxicillin-clavulanate (cholestatic) |
| Chronic Hepatic Injury | Methyldopa, Diclofenac (chronic hepatitis), Valproate, Amiodarone (steatosis) |
| Granulomatous Hepatitis | Allopurinol, Carbamazepine, Cephalexin |
| Chronic Cholestasis | Amitriptyline, Ampicillin, Chlorpromazine |
| Acute Steatosis | Amiodarone, Zidovudine, Herbal remedies |
| Mixed | Phenytoin |
| Cirrhosis | Methotrexate, Azathioprine, OCP |
| Phospholipodosis | Amiodarone, Chloroquine |
| Vascular Disease | OCP (hepatic vein thrombosis), Azathioprine, Vitamin A (SOS), Anabolic steroids, arsenic (Peliosis hepatis) |
| Vanishing Bile Duct Syndrome | Amoxicillin, Clindamycin, Carbamazepine |
| Biliary Sclerosis | Intra-arterial infusion 5-flurodexoyuridine |
Diagnosis involves detailed history, liver tests, and potentially liver biopsy. Temporal relationship between drug exposure and symptoms is key.
Treatment of DILI
Treatment is primarily drug withdrawal. Specific antidotes exist for acetaminophen (N-acetylcysteine) and valproic acid (L-carnitine) overdose. UDCA may be used in cholestatic DILI. Acute liver failure in DILI has high mortality and may require liver transplantation.
Extrahepatic Cholestasis: Obstructive Causes
Choledocholithiasis and Acute Ascending Cholangitis
Choledocholithiasis (CBD stones) affects 10-20% of symptomatic cholelithiasis patients. CBD stones can cause biliary obstruction and acute ascending cholangitis, a life-threatening condition. Biliary manipulation during ERCP is another common cause of cholangitis.
Clinical Features and Diagnosis
Choledocholithiasis can be asymptomatic. Symptomatic stones present with RUQ pain, fever, and jaundice (Charcot’s triad). Acute cholangitis pentad includes hypotension and altered mental status (less common). Elevated AP and GGT are seen in 90% of stone obstruction cases. Bile and blood cultures are often positive in cholangitis. TUS is initial screening, but MRCP and EUS are more accurate for CBD stone detection. ERCP is both diagnostic and therapeutic for stone retrieval.
Choledochal Cyst
Choledochal cysts are rare cystic dilatations of bile ducts, more common in Asians and females. Pancreatic secretion reflux into CBD damages bile ducts, leading to recurrent cholangitis, pancreatitis, and fibrosis.
Clinical Features and Diagnosis
Most cases are diagnosed in childhood. Adult presentation often lacks the classic triad of abdominal pain, jaundice, and palpable RUQ mass. TUS is initial imaging, but MRI/MRCP is preferred for biliary tree delineation. MRCP shows dilated CBD with saccular formation. Histology shows fibrous cyst walls, and adults may have biliary cirrhosis. CCA is a serious complication, occurring in ~20% of adult cases, making surgical resection recommended for most types.
Benign and Malignant Biliary Strictures
Benign biliary strictures can result from surgery, PSC, choledocholithiasis, chronic pancreatitis, trauma, and liver transplant. Malignant strictures are often caused by pancreatic cancer, CCA, gallbladder, and ampullary tumors. Klatskin tumors are CCA at the hepatic duct bifurcation. Strictures can be intraluminal or extrinsic.
Clinical Features and Diagnosis
Benign strictures may be asymptomatic or present with cholangitis. Pancreatic cancer typically presents with painless jaundice. Jaundice is also common in CCA. Weight loss and epigastric pain suggest malignancy. Extrahepatic cholestasis shows elevated AP and GGT with near-normal aminotransferases. Hyperbilirubinemia is often significant in pancreatic cancer. CA19-9 elevation suggests CCA in PSC patients. Imaging workup starts with TUS/CT, followed by MRCP/ERCP. ERCP brushings for cytology are crucial when malignancy is suspected, with advanced techniques enhancing CCA detection.
Management of Cholestasis
Pruritus Management
Pruritus is a challenging symptom in chronic cholestasis. Cholestyramine, a bile acid sequestrant, is first-line, but unpalatable and interferes with drug absorption. Rifampin reduces hepatic bile acid uptake and increases renal excretion, but liver tests must be monitored. Phenobarbital is less effective than rifampin and cholestyramine. Sertraline and paroxetine antidepressants may be helpful. Opioid receptor antagonists like naloxone, nalmefene, and naltrexone can provide relief but may cause opioid withdrawal. UDCA’s role is disease-specific. Plasmapheresis is reserved for refractory pruritus. Liver transplantation is an ultimate option for severe pruritus.
Fatigue Management
No consistently effective fatigue therapy exists. UDCA is not helpful, and fatigue may persist post-transplant. CNS stimulants like modafinil and low-dose amitriptyline may be beneficial in some patients.
Osteoporosis Management
Calcium and Vitamin D are recommended for all chronic cholestasis patients to prevent metabolic bone disease. Bisphosphonates are used for established osteoporosis, with alendronate being more effective than etidronate.
Fat-Soluble Vitamin Deficiency Management
Annual monitoring of vitamins A, D, E, K is recommended, with supplementation for deficiencies.
Hyperlipidemia Management
Cholestyramine’s role in hyperlipidemia management is unclear. UDCA can reduce total, VLDL, and LDL cholesterol. Clofibrate should be avoided as it paradoxically increases cholesterol in PBC. Statins can be used with liver enzyme monitoring, but may be avoided in severe cholestasis. Plasmapheresis is reserved for severe hypercholesterolemia to remove cholesterol and improve xanthomas and pruritus.
Outcome in Representative Case
The patient with IgG4-related autoimmune cholangitis responded to prednisone, achieving biochemical remission within months and maintaining normal liver tests on a low maintenance dose.
Conclusion
Differential diagnosis of raised alkaline phosphatase in the context of cholestasis requires a systematic approach, integrating history, physical examination, laboratory findings, and imaging. A step-by-step evaluation enables accurate diagnosis and tailored management for diverse cholestatic conditions.
References
[References from the original article, maintaining the original format and links]
Footnotes
Publisher’s Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Asma Siddique, Fellow in Hepatology.
Kris V. Kowdley, Center for Liver Disease, Virginia Mason Medical Center, Digestive Disease Institute, Seattle, WA.